ABSTRACTS FOR TALKS AND POSTERS
You can also click on ![]() left of the speaker's name in the program to display the corresponding abstract of main talks and short communications. For poster abstracts you have to browse through this page.
left of the speaker's name in the program to display the corresponding abstract of main talks and short communications. For poster abstracts you have to browse through this page.
CATEGORIES:
Generation and design of chimeric toxins
Directed enzyme prodrug therapy
Targeted RNases
Innovative tools to improve targeted tumor therapies
Targeted tumor therapies in clinical applications
Category: Generation and Design of Chimeric Toxins
TALK: Wednesday 01 April 2009, 12:00
Bispecific Ligand Directed Toxins for Solid Tumor Therapy
Daniel Vallera*, Seunguk Oh*
Therapeutic Radiology, University of Minnesota Cancer Center, Molecular Cancer Therapeutics, Minneapolis, MN 55455, USA
* These authors contributed equally to this work
The field of IT is limited by three major problems, potency, immunogenicity, and toxicity. To enhance potency and ability to kill solid tumors, we bio-engineered a new class of targeted toxins called bispecific ligand-directed toxins (BLT). BLTs are much more effective than their respective monospecific forms and work best because both targeting ligands are positioned on the same single chain (sc) molecule. For example, we cloned EGF4KDEL by positioning the cytokine human EGF next to human IL-4 on the same sc molecule with truncated PE. To reduce immunogenicity, the toxin was mutated to eliminate amino acids in 7 key epitopic regions that dictate B cell response. The resulting EGF4KDEL 7mut retained functional activity in entirety and had 80-90% reduced capacity to generate anti-toxin antibodies in normal mice. This represents an important step forward in the use of directed toxins in carcinoma therapy since using multiple injections of “reduced-immunogenicity BLT”, we were able to inhibit systemic breast cancer in a powerful dual reporter gene bioluminescence animal model. To reduce toxicity, we developed a unique systemic delivery approach based on blocking critical receptors on vital organs and studied it in a different pancreatic cancer model. In this model, a pre-dose of recombinant sc EGF13 (EGF-IL13) devoid of toxin was administered before DTEGF13 (DT390-EGF-IL13). This “toxicity blocking” approach permitted multiple treatment with doses exceeding MTD and resulted in significant systemic erosion of human pancreatic cancer in an orthotopic xenograft model.
New Targeted Therapies against Prostate Cancer with PSMA-specific Immunotoxins and Diabodies
Ursula Elsässer-Beile, Philipp Wolf, Patrick Bühler
Urology, University Hospital Freiburg, Experimental Urology, 79106 Freiburg, Germany
Prostate cancer is one of the most frequently diagnosed tumors. Since there are very limited treatment options for advanced disease, there is an increasing need for new treatment strategies. Prostate specific membrane antigen (PSMA) is an integral, non-shed membrane protein that is specifically expressed on prostate epithelial cells and strongly upregulated in prostate cancer. Therefore, it constitutes an ideal target for specific therapies. We constructed two recombinant immunotoxins, A5-PE40 and D7-PE40, consisting of the anti-PSMA scFv binding domains A5 and D7 as well as PE40 as toxin, a truncated form of Pseudomonas Exotoxin A. In vitro, a specific cytotoxicity of A5-PE40 and D7-PE40 on PSMA-expressing C4-2 prostate cancer cells could be measured. Treatment of C4-2 xenograft-bearing SCID mice caused a significant inhibition of tumor growth, whereas PSMA-negative DU145 tumors remained unaffected. The PSMAxCD3 diabody was constructed from an anti-CD3 scFv and A5. It could be shown in vitro that co-ligation of this diabody with CD4+ or CD8+ T-lymphocytes effectively eliminates PSMA-positive tumor cells. After incubation with diabody and target cells, T-lymphocytes showed an elevated expression of activation markers and the secretion of distinct cytokine profiles. The treatment of C4-2 xenograft-bearing SCID mice with the diabody and human peripheral blood lymphocytes proved to inhibit tumor growth significantly. Our preclinical results are promising and encourage further research on specific therapies against PSMA for treating prostate cancer.
Immunotoxins to Prostate Tumors
Marco Colombatti
Department of Pathology — Section of Immunology, Policlinico G.B. Rossi, University of Verona, 37134 Verona, Italy;
The concept of monotherapy in the management of tumor disease has been superseded in the past years by the use of more efficacious concomitant and/or sequential treatment protocols; in addition selective targeting of cytotoxic drugs to tumors may be more effective if several tumor-associated molecules are targeted because individual markers are not homogenously distributed at the surface of all tumor cells. Therefore eradication of the highest possible number of target cells may require combined treatments with immunotoxins directed to the different molecules. Prostate carcinoma (PCa) cells may express several different antigens at their surface: Prostate specific membrane antigen (PSMA), prostate stem cell antigen (PSCA), or epithelial cell adhesion molecule (EpCAM). To implement our idea of improving the efficacy of immunotoxin based therapy of PCa using different toxin vehicles recognizing different antigens, we have obtained immunotoxins (synthesized by chemical cross-linking or obtained by gene-fusion) to PSMA, PSCA and EpCAM. Their effects were investigated using prostate carcinoma cell lines as targets. Our data confirm that anti-PSMA immunotoxins are able to selectively kill PSMA+ cells (LNCaP and PSMA transfectants), immunotoxins to EpCAM can intoxicate not only LNCaP but also DU145 PCa cells and anti-PSCA IT are effective on PSCA+ cancer cells. Results on the properties and anti-tumor effects of immunotoxins to the different markers mentioned will be presented.
Immunokinases, a Novel Class of Immunotherapeutics for Targeted Cancer Treatment
Mehmet Tur1, Stefan Barth1, 2
1Chair of Applied Medical Engineering, Experimental Medicine and Immunotherapy, Helmholtz-Institute for Biomedical Engineering, University Hospital RWTH Aachen, 52074 Aachen, Germany; 2Pharmaceutical Product Development, Fraunhofer-Institute for Molecular Biology and Applied Ecology, 52074 Aachen, Germany
Certain characteristics of tumor cells make it possible to develop rational strategies for targeting tumors without harming normal cells. These include the presence of cell surface molecules that characterize the current state of the tumor and the genetic and epigenetic changes that activate oncogenes and inactivate tumor suppressor genes. Death-associated protein kinase 2 (DAPK2) is a calcium/calmodulin (CaM)-regulated pro-apoptotic serine/threonine kinase that acts as a tumor suppressor. Here we show that DAPK2 is downregulated in Hodgkin lymphoma-derived tumor cells and that promoter region hypermethylation is one mechanism for DAPK2 inactivation. To determine whether selective reconstitution of DAPK2 catalytic activity in these cells could induce apoptosis, we created a fusion protein comprising a human CD30 ligand conjugated to a human DAPK2 CaM-deletion mutant. Thus, recombinant immunokinase fusion protein DAPK2′-CD30L has a constitutive kinase activity with enhanced pro-apoptotic function. We show that this immunokinase induces apoptotic cell death specifically in CD30+/DAPK2-negative tumor cells in vitro and significantly prolonged overall survival in a disseminated Hodgkin lymphoma xenograft SCID mouse model. This proof-of-concept study provides the first demonstration of therapeutic strategies based on the restoration of a defective, tumor-suppressing kinase activity by a novel class of recombinant immunotherapeutics.
Targeted Induction of Apoptosis by Immunotoxin-like Fusion Proteins Employing Human Effector Functions
Winfried Wels, Hayat Mahmud, Robert Jabulowsky, Pranav Oberoi, Benjamin Dälken
Georg-Speyer-Haus, 60596 Frankfurt, Germany
Successful application in cancer patients has revived interest in recombinant immunotoxins as targeted therapeutics. These chimeric molecules combine specific recognition of cancer cells with selective delivery of a potent protein toxin of plant or bacterial origin. In our earlier work we have characterized Pseudomonas exotoxin A-based fusion proteins targeted to EGFR and the closely related ErbB2 (HER2) molecule. Elevated levels of these receptor tyrosine kinases have been found in many tumors of epithelial origin, and have been shown to contribute to cellular transformation. For an ErbB2-specific toxin, we could demonstrate safety and antitumoral activity in cancer patients. Nevertheless, repeated or prolonged treatment with such a molecule can be complicated by the development of neutralizing antibodies against the bacterial toxin domain. Hence, to reduce immunogenicity, we have generated similar immunotoxin-like fusion proteins employing apoptosis-inducing effectors of human origin. One such strategy is based on tumor-specific derivatives of the serine protease granzyme B (GrB) of cytotoxic lymphocytes. Recombinant GrB molecules that carry ligands specific for ErbB2 or EGFR were selectively internalized by tumor cells expressing the respective target receptors, and in the presence of an endosomolytic agent rapidly induced cell death. Ongoing work now aims at improvements in the molecular design of such fusion proteins, and the characterization of alternative human effector functions such as apoptosis inducing factor (AIF).
Development of Novel, Highly Cytotoxic Fusion Constructs Containing Granzyme B: Unique Mechanisms and Functions
Michael G. Rosenblum
M.D. Anderson Cancer Center, The University of Texas, Experimental Therapeutics, Houston, TX 77030, USA
Recombinant fusion proteins are an expanding, important class of novel therapeutic agents. The designs of these constructs typically involve a cell-targeting motif genetically fused to a highly toxic class of enzymes capable of ruthlessly attacking critical cellular machinery once delivered successfully to the cytoplasm of the target cell. Initial development of this class of constructs typically contained recombinant growth factors or single-chain antibodies as the cell-targeting motif fused to highly cytotoxic plant or bacterial toxins. This review describes second-generation molecules composed of cell-targeting molecules fused to highly cytotoxic human enzymes capable of generating intense apoptotic response once delivered to the cytoplasm. The human serine protease granzyme B has been shown to be extremely effective as a cytotoxic molecule when incorporated into numerous cell-targeting constructs. The biological activity of GrB-containing constructs rivals that of plant or bacterial toxins and appears to represent a new generation and class of completely human proteins with unique biological activities.
Tumor Targeting by Protease-Activated Anthrax Toxin Proteins
Stephen Leppla1, ShiHui Liu1, Randall Alfano2, Arthur Frankel2, Thomas Bugge3
1Laboratory of Bacterial Diseases, NIAID, NIH, Bethesda, MD 20817, USA; 2Cancer Research Institute, Scott & White Memorial Hospital, Temple, TX 76502, USA; 3Oral and Pharyngeal Cancer Branch, NIDCR, NIH, Bethesda, MD 20892, USA
Protein toxins have frequently had specificity for cancer cells conferred on them by attachment of polypeptides that bind to the tumor cell surface, e.g. antibodies. An alternative approach is to make these toxins dependent on tumor cell surface proteases such as matrix metalloproteinases (MMPs) and urokinase plasminogen activator (uPA). Anthrax toxin absolutely requires protease activation at the cell surface, a fact we have exploited to create agents having high specificity for tumors. Thus, anthrax protective antigen (PA) protein having the native furin cleavage site replaced by sequences cleaved by MMPs or uPA is able to specifically deliver the toxin’s lethal factor (LF) protease or LF fusion proteins into tumor cells. An LF fusion to Pseudomonas exotoxin A (FP59) has strong activity against xenographed tumors in mice when administered with PA having a uPA cleavage site (PA-U2). Current focus is on the combination of PA-L1, which depends on MMP activity, with native LF, which acts inside cells to cleave MAPKKs and inactivate key signaling pathways, including the pathway activated in BRAF-dependent tumors such as melanomas. The PA-L1 + LF combination is active on a range of transplanted tumors, and not only on melanoma. Mechanistic analyses show that this agent targets tumor endothelium and has a strong anti-angiogenic activity, suggesting it may have broad application for solid tumors. Rapidly accumulating knowledge on the structure and function of the anthrax toxin proteins promises that further improvements can be expected in the design of these anti-cancer agents.
G-01
POSTER
Characterization of a Recombinant Anti-PSMA Single-Chain Immunotoxin against Prostate Cancer
Philipp Wolf, Karen Alt, Patrick Bühler, Ulrich Wetterauer, Ursula Elsässer-Beile
Experimental Urology, University Hospital Freiburg, 79106 Freiburg, Germany
Prostate cancer remains a leading cause of cancer deaths in men. Whereas early disease can be successfully treated, no curative therapy exists for advanced stages. Therefore new agents for targeted treatment are urgently needed. The prostate specific membrane antigen (PSMA) is a cell surface glycoprotein that is specifically expressed on prostate epithelial cells and upregulated in all stages of prostate cancer. Therefore specific immunotherapy against this antigen may be a novel option for the management of this tumor. We constructed a recombinant immunotoxin, called A5-PE40, consisting of the single-chain antibody fragment (scFv) A5 against PSMA as the binding domain, and PE40, a truncated form of Pseudomonas Exotoxin A, as the toxin domain. In vitro, A5-PE40 showed a specific binding to PSMA positive cells of the human prostate cancer line C4-2. Furthermore, a high and specific cytotoxicity of the immunotoxin against C4-2 cells was measured with an IC50 value of 220 pM. In vivo, treatment of SCID mice bearing C4-2 xenografts with A5-PE40 caused a significant inhibition of tumor growth, whereas PSMA negative tumors remained totally unaffected. Due to its high and specific cytotoxicity and antitumorous activity the immunotoxin A5-PE40 represents a promising candidate for therapeutic intervention against prostate cancer.
G-02
POSTER
Co-Ligation of Prostate Cancer Cells with T-Cells by a PSMAxCD3 Diabody Leads to Lysis of the Tumor Cells
Patrick Bühler1, Eszter Molnar2, Philipp Wolf1, Ulrich Wetterauer1, Wolfgang Schamel2, Ursula Elsässer-Beile1
1Department of Urology, Experimental Urology, University Hospital Freiburg, 79106 Freiburg, Germany; 2Department of Molecular Immunology, Max-Planck-Institute for Immunobiology, 79108 Freiburg, Germany
Since there are currently no curative treatments for advanced prostate cancer, there is an urgent need for targeted therapies. Bispecific diabodies, redirecting T cells for the lysis of tumor cells have been shown to be potent tools in immunotherapeutic approaches. We have developed a PSMAxCD3 diabody against prostate cancer cells with two opposing binding sites: for the Prostate Specific Membrane Antigen (PSMA) and the T cell antigen receptor (TCR-CD3). This diabody was shown to effectively eliminate prostate cancer cells in vitro and in vivo. In the present study the mechanistic principles behind the controlled polyclonal T cell activation were to be further explored. The T cell mediated killing was strictly dependent on the co-incubation of the T cells together with PSMA-positive tumor target cells and could not be observed after co-incubation of T lymphocytes with PSMA-negative DU 145 cells. By measuring activation markers, proliferation and cytokine secretion it could be shown that after 24 h incubation with the diabody and the target cells both, CD4+ and CD8+ lymphocyte were activated. With respect to their cytotoxic activity, for both CD4+ and CD8+ cells the perforin granzyme pathway was the dominant mechanism. The presented PSMAxCD3 diabody can be regarded as suitable target-cell dependent agent for a controlled polyclonal T cell therapy of prostate cancer.
G-03
POSTER
Engineering of Bifunctional Anti-Eag1 Antibodies for Cancer Therapy
Franziska Hartung, Walter Stühmer, Luis A. Pardo
Department of Molecular Biology of Neuronal Signals, Max-Planck-Institute of Experimental Medicine, 37075 Göttingen, Germany
Antibody-based cancer therapy uses the high selectivity of antibodies to selectively destroy cancer cells. Therefore, a chief factor for an efficient antibody-based cancer therapy is the targeted antigen. We have selected the potassium channel Eag1 (ether a go-go) because this surface protein is not detected in normal tissues outside of the central nervous system, but more than 75% of tumors from different origins have been tested positive for Eag1 expression. We have generated specific anti-Eag1 monoclonal antibodies, but recombinant single chain antibodies (scFv) offer several advantages in comparison to whole antibodies, while retaining the full binding specificity because the antigen-binding surface is unaltered. scFv have better tissue penetration and are easier to produce or label than whole antibodies. We designed a scFv antibody which binds to the pore region of Eag1 from the extracellular side and fused it to different effectors like truncated Pseudomonas exotoxin A to generate an immunotoxin or an apoptosis-inducing ligand to mediate an antitumor immune response. The constructs are produced in a bacterial expression system in E. coli and purified using gel filtration and affinity chromatography. We currently test the efficacy of several antibody-based strategies for an effective targeting of cancer cells expressing Eag1.
G-04
POSTER
Anti PSCA Monoclonal Antibody and scFv-Fragment for Immunotherapy of Prostate, Pancreatic and Bladder Carcinoma
Giorgia Cremonese1, Giulio Fracasso1, Sara Cingarlini2, Cristina Anselmi1, Anita Boscaini1, Mariangela Figini3, Silvana Canevari3, Gianluigi Cetto2, Marco Colombatti1
1Department of Pathology — Section of Immunology, Policlinico G.B. Rossi, University of Verona, 37134 Verona, Italy; 2Unit of Medical Oncology, University of Verona, 37100 Verona, Italy; 3Department of Experimental Oncology, Istituto Nazionale Tumori, 20133 Milan, Italy
Unconjugated, toxin-conjugated or radiolabeled antibodies (Ab) recognizing tumor-associated antigens have proven beneficial for solid and hematolymphoid neoplasms. A suitable antigen for targeted therapy could be Prostate Stem Cell Antigen (PSCA), a cell surface-antigen expressed at low levels in normal prostate and over expressed in prostate, pancreatic and bladder carcinoma. Anti PSCA mAb was obtained by hybridoma technology and a recombinant version in single chain format (scFv) was generated by cloning the variable heavy (VH) and light chain (VL) sequences in the expression vector pHEN-2. mAb and scFv ability to recognize native PSCA was assessed by flow cytometry on SW780 (PSCA+) cells. The mAb demonstrated a good specificity and affinity, whereas the scFv showed a lower staining compared to whole mAb. The affinity/avidity of scFv was partially recovered making it divalent by cross-linking myc Tag by pre-incubation with anti myc-Ab. mAb was also able to recognize the glycosylated PSCA on prostate and pancreatic neoplastic tissue lysates by Western Blot. The antibody internalization assay demonstrated that the Ab-PSCA complex was internalized. This property was exploited for intracellular delivering of cytotoxic agents. The sequence coding for the Pseudomonas aeruginosa exotoxin A mutant PE40 was cloned at the 3′-end of the scFv yielding an anti-PSCA recombinant immunotoxin. Analysis of cytotoxic activity and specificity of scFv-PE40 are on-going.
G-05
POSTER
Induction of Programmed Cell Death in ErbB2/HER2-expressing Cancer Cells by Targeted Delivery of Apoptosis Inducing Factor (AIF)
Hayat Mahmud, Benjamin Dälken, Winfried S. Wels
Georg-Speyer-Haus, 60596 Frankfurt am Main, Germany
Apoptosis inducing factor (AIF) is a mitochondrial flavoprotein with NADH oxidase activity that has a vital function in healthy cells, but is also an important mediator of caspase-independent programmed cell death in stressed and damaged cells. Here we have generated a truncated AIF derivative (AIFΔ100), which lacks the mitochondrial import signal of the protein. Bacterially expressed AIFΔ100 was functionally active, and induced cell death upon microinjection into Vero cells accompanied by clear signs of apoptosis. For specific targeting to tumor cells, AIFΔ100 was genetically fused to the scFv(FRP5) antibody fragment which recognizes the ErbB2 (HER2) receptor tyrosine kinase frequently overexpressed in many human cancers. Recombinant scFv(FRP5)-AIFΔ100 (5-AIFΔ100) protein and a similar scFv(FRP5)-ETA252-366-AIFΔ100 (5-E-AIFΔ100) molecule harboring in addition the non-toxic translocation domain of Pseudomonas exotoxin A as an endosome escape function displayed binding to ErbB2-expressing cells followed by protein internalization and accumulation in intracellular vesicles. In the presence of the endosomolytic reagent chloroquine, 5-E-AIFΔ100 but not the similar 5-AIFΔ100 protein displayed potent cell killing activity, which was strictly dependent on the expression of ErbB2 on the target cell surface. Our results demonstrate that recombinant AIF specifically targeted to human cancer cells and delivered into the cytosol has potent cell-killing activity, suggesting this molecule as an effector function suitable for the development of humanized immunotoxin-like molecules.
POSTER and SHORT COMMUNICATION: Thursday 02 April 2009, 12:35
Tumor Targeting of Colorectal Carcinoma and Liver Metastasis with Shiga Toxin B
Matthias Maak1, Ludger Johannes2, Helmut Friess1, Klaus-Peter Janssen1
1Chirurgische Klinik und Poliklinik, Klinikum rechts der Isar der Technischen Universität München, 81675 Munich, Germany; 2CNRS-UMR 144, Institut Curie, 75248 Paris, France
Efficient methods for tumor targeting are eagerly awaited for metastasized colorectal cancer. A targeting vector should display molecular specificity, fast uptake into tumor cells, low immunogenicity, and the capacity to withstand inactivation. These requirements are satisfied by the non-toxic B-subunit of the bacterial Shiga toxin (STxB). The glycosphingolipid Gb3 (CD77), the receptor of Shiga toxin, is overexpressed on a wide range of tumors. Notably, we have shown that Gb3 is significantly increased in pancreatic and colorectal carcinoma, and in liver metastases. After Gb3 binding, STxB enters the cytoplasm via the retrograde route, bypassing the degrading environment of the lysosomes. This characteristic makes STxB a potential carrier for diagnostic and therapeutic purposes. Indeed, we could identify STxB as a promising tool for non-invasive and fibroscopic imaging, demonstrated by studies on genetic mouse models for colon cancer. Further analyses on short-term primary cell cultures of human colorectal cancer and liver metastases showed a fast intracellular STxB uptake. In contrast, benign adenomas with low expression of Gb3 showed neglectable uptake of STxB. Primary cultures are established from freshly collected tissue, thus avoiding the potential aberrations caused by extended periods of cell culture. Our current study focuses on the use of STxB for targeted therapy, coupled to SN38, a topoisomerase I inhibitor prodrug. This compound is being tested on primary cultures of colorectal carcinoma, as well as on xenograft models for non-invasive Micro-PET studies.
G-07
POSTER
Targeted Induction of Apoptosis in Tumor Cells by Chimeric Granzyme B Fusion Proteins
Robert Jabulowsky*, Pranav Oberoi*, Benjamin Dälken, Hayat Mahmud, Winfried S. Wels
Georg-Speyer-Haus, 60596 Frankfurt am Main, Germany
* These authors contributed equally to this work
Tumor cells are frequently insensitive to apoptotic stimuli due to deregulation of the apoptotic cell death pathways. Introducing into such cells a potent inducer that concurrently activates multiple targets within the apoptotic cascade might overcome apoptosis resistance and induce cell death. The serine protease granzyme B (GrB) of CTLs is able to enter target cells and rapidly induce apoptosis via caspase-dependent and caspase-independent mechanisms. Based on this broad activity, we are investigating recombinant GrB as an apoptosis-inducing effector function in chimeric molecules with potential therapeutic activity. Selectivity for tumor cells is contributed by fusing targeting domains to the enzyme that bind the epidermal growth factor receptor (EGFR) or the closely related ErbB2 (HER2/neu) protein, both overexpressed in various tumors of epithelial origin. We previously described prototypic GrB derivatives that carry an ErbB2-specific scFv antibody fragment or an EGFR-specific peptide ligand. These molecules were selectively internalized by tumor cells expressing the respective target receptors, and in the presence of an endosomolytic activity rapidly induced cell death. Our ongoing work aims at improving the design of such molecules to further enhance their selectivity and cytotoxicity. We modified the surface charge of GrB by site-directed mutagenesis to reduce intrinsic, non-specific cell binding of the GrB domain, which was observed in previous studies. The resulting GrB fusion proteins are being expressed in the yeast P. pastoris for functional analysis.
G-08
POSTER
Transferrin-targeted Saporin Induces Apoptosis in Glioblastoma Multiforme Cell Lines via Multiple Mechanisms
Annamaria Cimini1, Elisabetta Benedetti1, Sabrina Mei1, Giulio Laurenti1, Giuseppina Pitari1, Alessio Cialfi1, Francesco Giansanti1, Luana Di Leandro1, Maria Serena Fabbrini2, Rodolfo Ippoliti1
1Basic And Applied Biology, I-67100 , Italy; 2Istituto Di Biologia E Biotecnologia Agraria, Consiglio Nazionale Delle Ricerche, 20133 Milan, Italy
Glioblastoma multiforme (GBM) has been a focus of new therapy development because it is the most common primary brain tumor in adults. Notwithstanding standard-of-care therapy most patients develop tumor recurrence or progression. Thus testing new therapeutic approaches is mandatory. Since a variety of human brain tumors over-express the transferrin receptor we generated a chimeric toxin (T-S), which is composed of human transferrin and saporin, a plant type I RIP. We tested its activity on two different GBM cell lines, one expressing wt p53 (U87) and the other with inactivated p53 (GL15). T-S is highly toxic for both GBM cell lines and its activity is at least three orders of magnitude higher than that of the RIP alone. We studied the different pathways activated by T-S and free saporin in the two cell lines. The data obtained strongly point towards the activation of apoptosis via caspase 8 in GL15 cells and via caspase 9 in U87 cells. In both cell lines we noticed the activation of ERK1/2 pathways. Both these effects are not completely due to the catalytic activity of saporin, since a mutant saporin (KQ) lacking toxicity, elicited different response on the two cell lines tested, being U87 cell growth and viability inhibited by the treatment while GL15 being completely unaffected. These results suggest that saporin-based therapy may be affected by the cell molecular profile and a possible use of catalytically inactive saporin as a potential cytotoxic agent.
G-09
POSTER
The Potential Use of PDZ Domains in the Expression, Targeting and Activation of Modified Saporin Variants
Francesco Giansanti1, Luana Di Leandro1, Giuseppina Pitari1, Ilias Koutris1, Maria Serena Fabbrini2, Alessio Lombardi2, David J. Flavell3, Sopsamorn U. Flavell3, Stefano Gianni4, Rodolfo Ippoliti1
1Basic And Applied Biology, University of L'Aquila, I-67100, Italy; 2Istituto Di Biologia E Biotecnologia Agraria, Consiglio Nazionale Delle Ricerche, 20133 Milan, Italy; 3The Simon Flavell Leukaemia Research Laboratory, University of Southampton, SO16 6YD Southampton, United Kingdom; 4Biochemical Sciences, University Of Rome, I-00185 Rome, Italy
We describe an engineered form of the ribosome inactivating protein saporin (sap-VSAV), bearing a C-terminal extra sequence that is recognized and bound by the PDZ2 domain from the mouse PTP-BL protein. The binding of PDZ domain to saporin was expected to reduce saporin toxicity since the proximity of the catalytic site of the toxin to the C-terminus. The co-expression of sap-VSAV and PDZ2 genes in E. coli BL21 cells greatly enhances the production of the toxin in a soluble form. Surprisingly, the increase of production was not due to protection from bacterial intoxication due to saporin enzyme activity, but may arise from a stabilization effect of PDZ2 on the toxin molecule during biosynthesis. We found that once purified sap-VSAV is stable but is not toxic to free ribosomes, while it is fully active against human cancer cells. This strategy of co-expression of a toxin moiety and a soluble PDZ domain may represent a new system to increase the production of recombinant toxic proteins and suggest the selection of new extra sequences that could allow to target PDZ domains inside specific mammalian cellular domains. Furthermore these results indicate an activation mechanism of sap-VSAV, possibly by cellular proteases, before induction of toxicity.
G-10
POSTER
Recruitment of Natural Killer Cells for the Elimination of Tumor Cells
Sabine Bergelt, Hauke Lilie
Institute of Biotechnology, Martin-Luther-University Halle-Wittenberg, 06120 Halle, Germany
The function of Natural Killer cells (NK cells) in the context of the human immune system consists of the elimination of virus infected, several transformed and malign cells. This occurs upon molecular recognition of target cells by binding and activation of the activating receptor NKG2D on the surface of NK cells. Receptor binding leads to induced cytotoxic activity and elimination of target cells. With the help of an artificial bifunctional protein (B3-MICA), where a tumor-specific antibody fragment (B3) is coupled to a NKG2D-binding ligand (MICA), NK cells should be recruited to tumor cells and simultaneously be activated. Here, we describe the design and functional characterization of the bifunctional protein B3-MICA. The protein was generated by the coupling of MICA to the tumor-specific antibody fragment B3 via cysteine-containing polyionic peptides as heterodimerization motif. The resulting immunoconjugate B3-MICA could be shown to specifically activate NK cells, thus leading to the elimination of tumor cells.
POSTER and SHORT COMMUNICATION: Wednesday 01 April 2009, 12:55
Pichia Pastoris as a Host for Secretion of Saporin-based Chimeras with Anti-Cancer Potential
Maria Serena Fabbrini1, Alessio Lombardi1, Sara Bursomanno1, Teresa Lopardo1, Roberta Traini2, Pietro Della Cristina2, Marco Colombatti2, Rodolfo Ippoliti3, Sopsarmon Flavell4, Aldo Ceriotti1, David J. Flavell4
1Istituto Di Biologia E Biotecnologia Agraria, Consiglio Nazionale Delle Ricerche, 20133 Milan, Italy; 2Department of Pathology — Section of Immunology, Policlinico G.B. Rossi, University of Verona, 37134 Verona, Italy; 3Basic And Applied Biology, University of L'Aquila, I-67100, Italy; 4The Simon Flavell Leukaemia Research Laboratory, University of Southampton, SO16 6YD Southampton, United Kingdom
at the request of the author this abstract is only available in the printed version of the abstract book
Category: Directed Enzyme Prodrug Therapy
TALK: Thursday 02 April 2009, 14:30
The ADEPT Story so far
Kenneth D. Bagshawe
Medical Oncology, Imperial College London, Charing Cross Campus, W6 8RF London, United Kingdom
Antibody directed enzyme prodrug therapy (ADEPT) has the potential to generate a high concentration of cytotoxic drug at cancer sites and to restrict that action to cancers. Pre-clinical studies in xenografted nude mice have been invaluable but are not infallable and it is only through clinical studies that progress can be made. An understanding of the pharmacokinetics of each component of the system is emphasized in this presentation. Although it is 20 years since this approach was first proposed and encouraging results obtained in advanced colorectal cancer, the remaining obstacles to fulfilling its potential can now be defined.
TALK: Thursday 02 April 2009, 15:15
Recombinant Fusion Proteins for Targeted Enzyme Prodrug Therapy in Colon and Pancreatic Cancer
P. Markus Deckert1, Ulf Petrausch2, Vânia Coelho3, Hossein Panjideh4
1Innere Klinik II, Klinikum Brandenburg, 14770 Brandenburg an der Havel, Germany; 2Universitätsspital Zürich,
Klinik für Onkologie, 8091 Zürich, Switzerland; 3University of Southampton, School of Medicine - Cancer Sciences, Southampton, Hampshire,
SO16 7PX, United Kingdom; 4Max-Delbrück-Center for Molecular Medicine, Dept. Molecular Tumor Genetics and Immunogenetics, 13125 Berlin, Germany
Antibody-directed enzyme-prodrug therapy (ADEPT) utilizes antibody-enzyme constructs for targeted enzyme delivery to tumors and subsequent localized prodrug activation. Its potential has been demonstrated in clinical studies. Its requirements for the antibody-enzyme construct are demanding, though: The enzyme must be of non-human origin, but should not be immunogenic. The complete protein must not be too large in order to allow for efficient tumor penetration. The antibody-enzyme construct must be of a defined structure and quality — and should be produced at an economically viable scale. Recombinant antibody-enzyme fusion proteins and deimmunization-techniques such as polyethylene glycol (PEG) conjugation promise to solve most of these problems. Here, we introduce our work in developing fusion proteins for ADEPT based on single chain variable-chain fragments (scFv) of various antibodies and yeast cytosine deaminase (CD), which converts the prodrug 5-fluorocytosine into 5-fluorouracil. We have developed an ADEPT system based on the gpA33 antigen of colon and pancreatic cancers, generating various A33scFv::CD fusion constructs. After a number of methodological problems in fusion protein expression we could finally demonstrate the efficacy of this ADEPT system in vivo. Additional fusion proteins for other antigens are under development and will be introduced as well as a pilot study on PEGylation of antibodies. In summary, we conclude that the development of antibody-enzyme constructs meeting the requirements for ADEPT as well as the regulatory ones for future drugs is feasible.
TALK: Thursday 02 April 2009, 15:50
Bench to Bedside with Recombinant Antibody-based Cancer Therapeutics
Kerry Chester, Berend Tolner, Surinder Sharma, Heide Kogelberg, Barbara Pedley, Astrid Mayer, Roslyn Francis, Duncan Wilkins, Geoff Boxer, Alan Green, John Hartley, Helen Lowe, Chris Taylorson, Richard Begent
Department of Oncology, UCL Cancer Institute, Paul O'Gorman Building, University College London, 72 Huntley Street, London WC1E 6BT, United Kingdom
The presentation will address the design and production of recombinant antibody fusion proteins for cancer treatment. In vitro and in vivo examples of therapeutics built from single chain Fv antibody fragments (scFv) will be discussed. The bench to bedside process, including manufacture to standards of good manufacturing practice (GMP), is exemplified with a case study of a fusion protein developed for antibody directed enzyme prodrug therapy (ADEPT). ADEPT is a staged therapy: a tumor-selective antibody is used to target an enzyme to cancerous deposits and a subsequently administered prodrug is activated by the targeted enzyme to generate a potent cytotoxic in the tumor mass. ADEPT requires highly selective delivery of enzyme to tumor. We have achieved this by molecular design and generation of a multifunctional genetic fusion protein of CPG2, a bacterial enzyme, with MFE-23, an scFv directed to carcinoembryonic antigen. The fusion protein (MFE-CP) is effective in pre-clinical ADEPT systems and can be manufactured to GMP in Pichia pastoris for use in patients. Phase I/II trials of ADEPT using MFE-CP show evidence of efficacy in patients with advanced disease. These trials illustrate the potential of multifunctional targeted proteins in cancer therapy and provide a platform of understanding for future applications. MFE-CP is immunogenic and a new ADEPT system, based on a humanized version of MFE-23 and a mutated human enzyme, is in pre-clinical development.
D-01
Synthesis and Biological Evaluation of New Duocarmycin Prodrugs for a Selective Cancer Therapy
Frank Behrendt, Michael Müller, Kianga Schmuck
Institut für Organische und Biomolekulare Chemie, Universität Göttingen, 37077 Göttingen, Germany
Chemotherapy of malign tumors is generally accompanied by serious side effects as selectivity of common anti-cancer drugs is unsatisfying. A promising method for a selective treatment of cancer is the ADEPT approach (Antibody Directed Enzyme Prodrug Therapy) [1]. During the last years we have developed prodrugs showing an excellent selectivity with an QIC50 (= IC50 of prodrug / IC50 of prodrug and cleaving enzyme) of about 5000 [2–4]. In contrast to the binary ADEPT concept, for the Prodrug Monotherapy (PMT) an immunoconjugate is not necessary, since the detoxifying unit of the prodrug can be cleaved either by enzymes occurring in a higher concentration in the tumor tissue or as a new approach by laser light. In order to investigate the prodrug's uptake into the cell or cell nucleus it can be labeled with a fluorescent dye and localized by fluorescence microscopy. Accordingly, novel seco-CBI-DMAI prodrugs 2 based on the cytotoxic antibiotic Duocarmycin SA 1 were synthesized in enantiopure form [4] and will be analyzed by cell culture and animal assays.
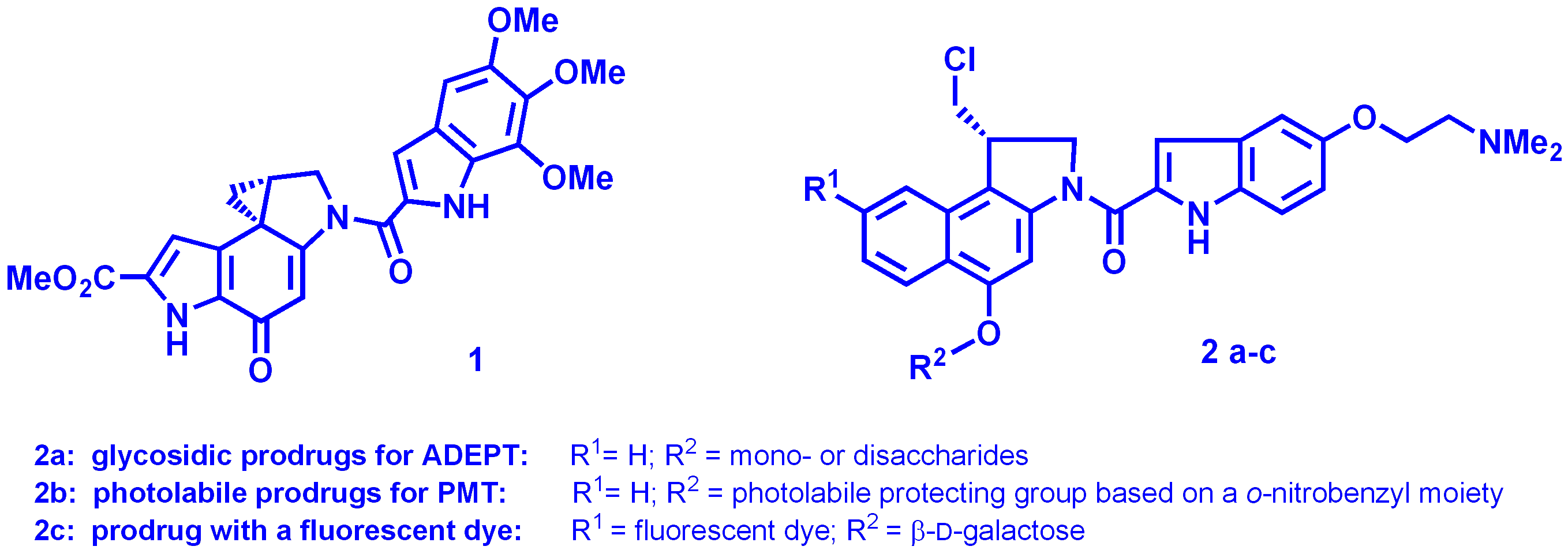
For ADEPT, new glycosidic prodrugs 2a with either a mono- or disaccharide moiety were prepared. The latter compounds are expected to have a better water solubility and a higher QIC50 than the glycosides of monosaccharides. For PMT using laser light, a UV-A-photoactivatable prodrug 2b was synthesized. Moreover, photolabile groups are under development which can be cleaved with a frequency doubled Nd:YAG-Laser being used for surgery. For fluorescence microscopic investigations the prodrug 2c with the dye 5-SFX (λmax,abs. = 486 nm, λmax,em. = 526 nm) at C-8 was synthesized.
References:
[1] Bagshawe KD, Br J Cancer 56:531-532 (1987).
[2] Tietze LF, Major F, Schuberth I, Angew Chem 118:6724-6727 (2006).
[3] Tietze LF, Major F, Schuberth I, Angew Chem Int Ed 45:6574-6577 (2006).
[4] Tietze LF, Schuster HJ, Schmuck K, Schuberth I, Alves F, Bioorg Med Chem 16:6312–6318 (2008).
[5] Tietze LF, von Hof JM, Krewer B, Müller M, Major F, Schuster HJ, Schuberth I, ChemMedChem 3:1946-1955 (2008).
D-02
Targeted Tumor Therapy with the Recombinant Fusion Protein A33scFv::CDy
Nicole Schellmann1, Hossein Panjideh1, 3, Christopher Bachran1, P. Markus Deckert2, Hendrik Fuchs1
1Zentralinstitut für Laboratoriumsmedizin und Pathobiochemie, Charité — Universitätsmedizin Berlin, 12200 Berlin, Germany; 2Innere Klinik II, Klinikum Brandenburg, 14770 Brandenburg an der Havel, Germany; 3Present address: Max-Delbrück-Center for Molecular Medicine, Dept. Molecular Tumor Genetics and Immunogenetics, 13125 Berlin, Germany
The antibody-directed enzyme prodrug therapy (ADEPT) enables a targeted tumor therapy with protein constructs containing a prodrug-converting enzyme and a targeting molecule. The recombinant fusion protein A33scFv::CDy combines the specific antigen binding and enzymatic properties of scFv A33 and cytosine deaminase (CD), respectively. CD converts the prodrug 5-fluorocytosine into 5-fluorouracil (5-FU), which is used in conventional chemotherapy. The specific antigen binding of scFv A33 prevents a systemic toxicity and other side effects of 5-FU. The antigen A33 is a suitable target for tumor therapy since it is expressed on almost all colon cancer cells (> 95 %). We performed biodistribution studies with radiolabeled A33scFv::CDy and revealed successful in vivo tumor targeting, which served as a basis for further investigations on the therapeutic efficacy of A33scFv::CDy for ADEPT. For the therapeutic in vivo experiments nude mice were injected with A33-expressing human colon carcinoma cells (LIM1215). After initial tumor growth the adjunction of A33scFv::CDy and 5-FC resulted in reduced tumor growth. Thereby we reported a successful application of ADEPT in vivo for the treatment of A33-positive colon carcinoma cells. Our data is promising and presents ADEPT as a suitable therapeutic strategy for the treatment of colon carcinomas.
TALK: Thursday 02 April 2009, 09:15
ONCONASE® (ranpirnase) as a Cancer Drug and Targeted Payload
Kuslima Shogen
Alfacell Corporation, CEO & Founding Scientist, Somerset, NJ 08873, USA
![]()
ONCONASE (ONC) causes RNA damage in non-coding RNAs (e.g. tRNA, microRNAs) as a prerequisite for tumor cell death. Moreover, ONC can increase sensitivity to other therapeutics. Targeting pathways regulated by NFκB, for example, can reverse chemoresistance to doxorubicin. More than 950 patients have been treated with ONC as a single agent and in combination drug therapy. The favorable safety profile allowed pharmacologically therapeutic doses to be achieved. Results of a recently completed Phase IIIb clinical trial for patients with unresectable malignant mesothelioma (Protocol P30-302, Part III) demonstrated real responses and clear clinical benefit with low incident of toxicity and no drug related deaths. Results were significant in patients who failed previous treatments. ONC significantly extended median survival when combined with doxorubicin (10.5 months, p=0.033) compared to doxorubicin alone (9 months). Long-term survival was also better in the patients that received ONC along with doxorubicin (1 year, 43% vs. 33% and 2 year, 23% vs. 10% survival rates). This outcome has a basis in preclinical studies for treating resistant tumors. Taken together, the extensive body of preclinical and clinical work showing anticancer effects of ONC provides an important rationale for the use of ONC as a payload in antibody targeted cancer strategies. It is expected that extending the profile of ONC responsive tumors and improving the effect of therapeutic antibodies could significantly increase the current options of anticancer treatments.
TALK: Thursday 02 April 2009, 09:50
Integrating Extracellular and Intracellular Specificity
Susanna Rybak
Bionanomics, LLC, #3036, Green Cove Springs, FL 32043, USA
ONCONASE® (ONC; ranpirnase) has been evaluated in clinical trials for a variety of cancers including unresectable malignant mesothelioma. Results show that ONC enhances the clinical benefit of standard chemotherapeutics even in drug resistant patients. These results likely relate to intracellular genes and pathways activated by ONC. Expression profiling using microarrays reveals that hundreds of genes are regulated by ONC in human malignant mesothelioma and breast cancer cell lines. The tumor suppressor gene IL24 is markedly up-regulated in mesothelioma cells as are genes encoding members of the GADD family and components of the MAPK signaling pathway in both mesothelioma and breast cancer cells. The known selective apoptosis of tumor cells by IL24 as well as the importance of GADD and MAPK to oncogene driven tumor growth may contribute to selective intracellular anticancer effects of ONC. Conjugating ONC to agents that recognize extracellular tumor associated molecules further increases its potency and specificity. ONC conjugates were directed to three different tumor associated molecules that targeted the transferrin receptor, CD22 antigen and the P-glycoprotein multidrug resistance transporter. These antibody-targeted ONC conjugates were effective in preclinical models, caused little non-specific toxicities in mice and displayed favorable formulation properties. Understanding the reasons for their potencies coupled with elucidating the intracellular events that contribute to tumor cell specificity will lead to improved variations of targeted ONC.
TALK: Thursday 02 April 2009, 10:50
Development of ImmunoRNases for Treatment of CD22+ Malignancies
Michaela Arndt1, Athanasios Mavratzas1, Ludger Grosse-Hovest2, Evelyn Exner1, Tamana Karimi1, Iduna Fichter3, Susanna Rybak4, Kuslima Shogen5, Gottfried Brem6, Dirk Jäger1, Jürgen Krauss1
1National Center for Tumor Diseases (NCT), University of Heidelberg, 69120 Heidelberg, Germany; 2Department of Immunology, Eberhard Karls University Tübing, 72076 Tübingen, Germany; 3Experimental Pharmacology, Max Delbrück Center for Molecular Medicine, 13125 Berlin, Germany; 4Bionanomics, LLC, #3036, Green Cove Springs, FL 32043, USA; 5Alfacell Corporation, Somerset, NJ 08873, USA; 6Agrobiogen GmbH, 86567 Hilgertshausen, Germany
For targeting CD22 malignancies we have previously generated a highly stable, humanized single chain Fv (scFv) fragment from the rapidly internalizing anti-CD22 monoclonal antibody RFB4. To further develop this antibody into therapeutically valuable reagents, we have converted the humanized RFB4 scFv into a humanized diabody and IgG1, respectively. In order to create novel anti-CD22 immunoRNases, this set of humanized antibodies was used for genetic fusion or chemical conjugation to human angiogenin (ANG) or amphibian Onconase (Onc), respectively. We show that — as a key requirement for further clinical development of these compounds — even structurally highly complex dimeric immunoRNase fusion proteins can be produced from stably transfected mammalian cell lines and purified from cell culture supernatants to homogeneity. Furthermore, we for the first time demonstrate the proof-of-concept that catalytically active dimeric immunoRNase fusion proteins are likewise producible in the serum of transgenic rabbits in high yield without mediating any toxic effects to the animals. Experiments for evaluating the pharmacokinetics and efficacy of the novel anti-CD22 immunoRNases in tumor-bearing immunodeficient mice are currently ongoing.
TALK: Thursday 02 April 2009, 11:25
Human RNase Fusion Proteins for Tumor Therapy
Stefan Dübel1, Christian Menzel1, Thomas Schirrmann1, Michael Hust1, Zoltan Konthur2, Thomas Jostock1
1Dept. Biotechnology, Institut für Biochemie und Biotechnologie, 38106 Braunschweig, Germany; 2Max Planck Institute of Molecular Genetics, 14195 Berlin, Germany
With increasing numbers of human antibodies entering the clinics, the problem of generating human antibodies to any desired specificity is solved. For example, by using phage display and our Hyperphage technology, we have set up a proven and robust structure for the in vitro generation of human antibody fragments. The development of antibody fusion proteins offers the perspective to improve the efficacy of naïve IgG for tumor therapy by adding novel effector functions, which would allow different therapeutic paradigms. This approach, however was hampered for decades by the lack of human fusion partners as effectors. Many immunotoxins have been designed and tested, but all to often caused severe side effects due to their immunogenicity and/or unspecific toxicity. Further, their production frequently requires refolding steps. Here, human effector domains may be employed to avoid the disadvantages. A now well established class of effectors are RNases. They are usually non-toxic while in circulation but highly effective in cell killing after internalization. We engineered and successfully produced in mammalian cells an entirely human immunoenzyme directed against CD30+ lymphomas. It was constructed from an scFv-Fc antibody fragment and a human RNase. It did not affect the human embryonal kidney cells used for its production by secretion into the supernatant, but strongly inhibited proliferation of CD30+ lymphoma cells with an IC50 = 3.3 nM.
References:
Menzel C, Schirrmann T, Konthur Z, Jostock T, Dübel S. Blood 111:3830-3837 (2008).
TALK: Thursday 02 April 2009, 12:00
Novel Human Anti-ErbB2 Immunoagents
Claudia De Lorenzo, Fulvia Troise, Gennaro Riccio, Paolo Laccetti, Giuseppe D'Alessio
Dept. of Structural and Functional Biology, University of Napoli Federico II, 80126 Napoli, Italy
Overexpression of ErbB2 receptor is associated with progression of malignancy of breast cancer. Herceptin, a humanized anti-ErbB2 antibody, has proved to be effective in the immunotherapy of breast carcinoma. However, it can engender cardiotoxicity and a high fraction of breast cancer patients are resistant to Herceptin-treatment. Two novel human anti-tumor immunoconjugates were engineered in our laboratory by fusion of a human anti-ErbB2 scFv (Erbicin) with either a human RNase or the Fc region of a human IgG1, called Erb-hRNase and Erb-hcAb (human anti-ErbB2-compact Antibody), respectively. The Erbicin-derived immunoagents (EDIA) target an ErbB2 epitope different from that of Herceptin, and are selectively cytotoxic for ErbB2-positive cancer cells in vitro and in vivo. We report that EDIA did not show cardiotoxic effects both in vitro on rat cardiomyocytes and in vivo on a mouse model, whereas Herceptin was strongly toxic. This difference was found to be due to their different effects: Herceptin, at difference with Erb-hcAb, induces apoptosis in cardiac cells. More interestingly, we found that EDIA are active on Herceptin-resistant cells both in vitro and in vivo. The sensitivity of these cells to treatment with EDIA is likely due to the different epitope recognized by them, since Erb-hcAb, at difference with Herceptin, was able to inhibit the signaling pathway downstream ErbB2. These results suggest that EDIA could fulfill the therapeutic need of patients either ineligible to Herceptin treatment due to cardiac dysfunction, or resistant to Herceptin.
Category: Innovative Tools to Improve Targeted Tumor Therapies
TALK: Wednesday 01 April 2009, 17:45
Red Blood Cells as Carriers for Targeted Drug Delivery to Tumor Cells
Hans Bäumler
Berlin Brandenburg Center for Regenerative Therapies (BCRT), Charité — Universitätsmedizin Berlin, 10117 Berlin, Germany
In an attempt to reduce main drawbacks of chemotherapy with anti-cancer agents drug delivery systems have been introduced that can deliver these drugs at the site of interest. The red blood cells (RBC) represent a potential system to carry drugs to the desired site of therapeutic action and provide an extraordinary vehicle for the dissemination of drugs in the circulation, due to the ability of their membranes to be opened and resealed. This carrier system is biocompatible, non-immunogenic, has a long life span and a large capacity. We developed a method for loading RBC with nanoparticles (NP) (size 10-80nm). Model drugs are 5-FU and FUAC, respectively. Additionally loading the RBC with magnetite NPs and magnetically focusing them to the desired sites or coupling of specific antibodies to the surface of RBC or both can realize targeting of organs or tissues. Magnetite loaded RBC can be visualized by MRI, offering the opportunity for diagnostic monitoring of the therapy. RBC carriers cannot just deliver a high dosage of different drugs but also protects them from inactivating effects and minimizes side reactions. Liver tumor or circulating tumor cells are first targets, which can be treated using the carriers. The uptake of RBC by tumor cells works in vitro as well as in vivo (animal experiments).
Acknowledgment: The author wants to acknowledge the contributions of his colleagues R. Georgieva, A. Müller, M. Müller, N. Sternberg, Z. Abdalah, S. John, U. Teichgräber, B. Paulke, D. Wang, M. Chanana, N. Buske and the financial support of the EU (EFRE-ProFIT-10134275).
TALK: Wednesday 01 April 2009, 18:20
Manipulation of Other Processes to Improve the Effectiveness of Tumor-targeted Toxins
Andrew Thorburn
Pharmacology, University of Colorado Denver, Aurora, CO 80010, USA
Tumor-targeted toxins kill cells by interfering with fundamental cellular activities such as protein synthesis and one might therefore expect that all cells targeted would be killed by similar mechanisms when this happens. However, we found that an EGFR-targeted diphtheria toxin (DT-EGF) kills glioblastoma cells by a caspase-independent mechanism that involves high levels of autophagy, which inhibits cell death by blocking apoptosis. In contrast, DT-EGF kills epithelial tumor cells by caspase-dependent apoptosis and in these cells autophagy is not induced. These differences allowed us to discover that the different death mechanisms were associated with differences in the release of an immuno-modulator protein (HMGB1) and that autophagy induction is required and sufficient to cause release of HMGB1 from the dying cells after treatment with DT-EGF. Here I will discuss the implications of these findings — in particular whether or not we should attempt to manipulate autophagy while treating tumor cells with targeted toxins.
TALK: Wednesday 01 April 2009, 18:55
Cytochrome P450 Specific Activation of Prodrug Ultrapotent Chemotherapeutics
Mark Sutherland, Klaus Pors, Jason Gill, Paul Loadman, Steve Shnyder, Laurence H Patterson
Institute of Cancer Therapeutics, University of Bradford, BD7 1DP Bradford, United Kingdom
Cytochrome P450's (CYPs) are a superfamily of mixed function oxidases responsible for metabolising and detoxifying xenobiotics in vivo. Elevated expression of several CYPs has been demonstrated in human cancers, with negative or low expression observed in the corresponding normal tissue. Exploitation of this differential has been suggested for the development of novel anticancer agents dependent upon CYP-mediated oxidation for conversion from an inactive to a potent chemotherapeutic. Pyrolloindoles are a family of ultrapotent natural agents with the ability to inhibit DNA replication and induce cell death, a process mediated through a specific hydroxyl group on the molecule. However these agents are not tumor-selective and can induce off-target toxicities. Therefore, pyrolloindole prodrugs activated to the potent agent through CYP-mediated hydroxylation should result in tumor-selective and potent chemotherapeutics. We have synthesized a series of chloromethylindoline prodrugs lacking the critical hydroxyl group, targeted at activation by CYP1A1. Metabolism studies using bactosomes, liver homogenates and selective inhibitors supported the CYP-selective activation of these agents. Several of these agents were shown to be ultrapotent (low nM activity) against CYP1A1 expressing cell lines in vitro. In addition, the prodrug is well tolerated in vivo with no observable systemic toxicity and inducing a significant retardation of tumor xenograft growth. Taken together, these data provide proof of concept for the development of CYP-mediated tumor-selective chemotherapeutics.
TALK: Thursday 02 April 2009, 08:30
Convection Enhanced Delivery: Neurosurgical Issues
Walter A. Hall
Neurological Surgery, Chairman, 612 Jacobsen Hall, Syracuse, NY 13210, USA
Because primary brain tumors have a poor prognosis, this has led investigators to develop, new innovative therapies such as targeted toxins. These large molecules do not cross the blood brain barrier and must be delivered into the brain by a technique known as convection enhanced delivery (CED). When administering these agents, there are a number of pharmacokinetic considerations that must be considered that will directly affect the volume of distribution of the drug and its the therapeutic effect. A number of different catheter types have been used to perform CED with a hollow fiber design offering several advantages over other variations. Specific parameters have been developed to optimize the placement of the catheters in order to enhance drug distribution in the brain. Considerable effort have been expended to identify a reliable way to image the distribution of targeted toxins administered by CED using magnetic resonance imaging and single photon emission computed tomography. Unfortunately many infusions performed in tumor patients are unsuccessful due to ventricular/subarachnoid leak or pooling of the drug in necrotic tumor tissue. To date, no targeted toxin clinical trial has demonstrated statistically significant clinical results leading to the universal acceptance of this treatment. Other agents such as standard chemotherapy or liposomal preparations have been delivered by CED. Non-neoplastic neurological diseases are being considered for treatment by CED with and treating different locations of the brain other that the cerebral hemispheres are under investigation.
TALK: Friday 03 April 2009, 08:30
Reducing the Immunogenicity of the Pseudomonas Exotoxin-based Immunotoxins
Masanori Onda, Ira Pastan
Center for Cancer Research, NIH/NCI, Laboratory of Molecular Biology, Bethesda, MD 20892-4264, USA
Recombinant ITs consisting of an Fv fragment joined to a truncated form of Pseudomonas exotoxin A (termed PE38) have been evaluated in clinical trials for the treatment of various cancers. IT BL22 contains the Fv of anti-CD22 antibody (Ab) fused to PE38. BL22 has produced many complete remissions in drug resistant hairy cell leukemia (HCL). In patients with hematological malignancies, the immune system is suppressed and ITs can be given for many cycles increasing their efficacy. But in patients with solid tumors, a neutralizing Ab response to PE38 prevents repeated treatments to maximize the benefit. There is evidence that human and mouse Abs recognize similar epitopes on foreign proteins such as PE38. Therefore we analyzed the murine Ab response as a model to identify the B cell epitopes associated with PE38. Sixty distinct MAbs to PE38 were characterized. Mutual competitive binding of the MAbs indicated the presence of 7 major epitope groups and 13 subgroups. The location of each epitope on PE38 was determined by preparing single amino acid mutants of PE38 in which bulky surface residues were mutated. Mutations, which did not bind to Ab, were judged to have lost the epitope, because we identified single point mutations to destroy each epitope structure. To allow more treatment cycles, we have produced a less immunogenic IT by combined 8 mutations that eliminate most epitopes. The new IT is less immunogenic in mice, yet retain full cytotoxic activity. Modification of B cell epitopes is a practical approach to the production of less immunogenic protein therapeutics.
TALK: Friday 03 April 2009, 09:15
Using Internalizing Cell Surface Receptors to Target Eukaryotic Elongation Factor 2
Stefan Barth1, 2, Mehmet Tur1
1Experimental Medicine and Immunotherapy, Chair of Applied Medical Engineering, Helmholtz-Institute for Biomedical Engineering, University Hospital RWTH, 52074 Aachen, Germany; 2Pharmaceutical Product Development, Fraunhofer-Institute for Molecular Biology and Applied Ecology, 52074 Aachen, Germany
Eukaryotic elongation factor 2 (EEF2), a member of the GTP hydrolase superfamily, is a cytoplasmic factor essential for protein biosynthesis. Inhibition of EEF2 by e.g. bacterial or plant-derived cytotoxic enzymes is known to arrest protein synthesis and induce apoptosis. As aptamers selectively binding to internalized tumor-associated antigens can be used as a nucleic acid-based carrier to deliver cytotoxic siRNAs, we used an aptamer binding to PSMA, a cell surface glycoprotein found on prostate cancer cells and joined its 3′ end to a siRNA specific for EEF2 mRNA. In order to enhance therapeutic efficacy of the transcript, we increased the valency of the construct by rational design. Here we will present cell surface receptor-specific delivery of functional aptamer-siRNA transcripts to PSMA-expressing prostate cancer cells as well as specific cytotoxicity resulting from siRNA-induced silencing of EEF2. Increasing the valency of the aptamer resulted in enhanced cytotoxicity compared to the monovalent constructs. Fluorescently labeled monovalent and bivalent aptamer-siRNA transcripts bound selectively to PSMA-expressing tumor cells and showed the same intensity of fluorescence. However, the bivalent transcripts showed significantly enhanced cytotoxicities with IC50 values in the range of 0.39 ± 0.07 µM and 0.17 ± 0.04 µM assayed in a dose-dependent manner compared to the monovalent construct (>0.8 µM). The results presented will be demonstrating the usefulness of aptamer-based delivery molecules as a novel class of siRNA-based therapeutics.
TALK: Friday 03 April 2009, 09:50
Development of Liposomal siRNA (AtuPLEX) for RNAi-mediated Therapeutic Applications
Ansgar Santel
Silence Therapeutics AG, 13125 Berlin, Germany
![]()
Functional RNAi in vivo requires the functional delivery of siRNA to the cytoplasm of the desired cell type within the body. Consequently, delivery remains the main obstacle for the development of RNAi-based therapeutics. For functional uptake of siRNA molecules, appropriate formulation strategies might facilitate the siRNA to overcome several cellular barriers in vivo and confer pharmacological properties. We have developed a cationic liposomal siRNA formulation (siRNA-lipoplex/AtuPLEX) for “systemic RNAi”. The AtuPLEX (liposomal siRNA) can be used for RNAi in the vascular endothelium, when applied intravenously. Therefore, an RNAi-mediated modulation of the vascular endothelium in vivo would offer many opportunities for developing new therapeutic interventions. The AtuPLEX technology is the basis of our current therapeutic program Atu027 in oncology. Pre-clinical research data will be discussed and an update on the status of our current drug candidate Atu027 will be provided.
TALK: Friday 03 April 2009, 10:50
Saponins as Tool for Drastically Improved Targeted Tumor Therapy
Hendrik Fuchs1, Diana Bachran1, Horst Dürkop2, Mark Sutherland3, Alexander Weng4, Romy Urban1, Stefanie Schneider1, Christian Müller1, Matthias Melzig4, Christopher Bachran1
1Zentralinstitut für Laboratoriumsmedizin und Pathobiochemie, Charité - Universitätsmedizin Berlin, 12200 Berlin, Germany; 2Institut für Pathologie, Charité - Campus Benjamin Franklin, 12200 Berlin, Germany; 3Institute of Cancer Therapeutics, University of Bradford, BD7 1DP Bradford, United Kingdom; 4Institute of Pharmacy, Free University Berlin, 14195 Berlin, Germany
Treatment of tumors with cytotoxic agents coupled to antibodies or ligands directed to tumor cell-specific structures is promising as demonstrated in numerous clinical studies and by approved drugs such as Mylotarg and Ontak, however, side effects are often severe and efficacy is limited due to low penetration of solid tumors and moderate cellular drug uptake. Moreover, production of antibodies is expensive and further restricts broad application. Here we describe the combined application of a glycosylated triterpenoid composite (Spn) and epidermal growth factor receptor (EGFR)-targeted chimeric toxins (SA2E). The cytotoxicity of SA2E on murine TSA tumor cells transfected with human EGFR was enhanced 20,000-fold by low non-permeabilizing Spn concentrations in a synergistic manner. Application in BALB/c mice bearing a solid TSA-EGFR tumor resulted in 94% tumor volume reduction with a 50-fold lower chimeric toxin concentration compared to pure SA2E treatment. Side effects as monitored by observable complications, body weight, blood parameters, histological analyses and antibody responses were only moderate and usually reversible. Since the required toxin amount is only 2% of that employed in conventional targeted therapies, the presented treatment is not only favorable with respect to tumor therapy but also in economical terms.
TALK: Friday 03 April 2009, 11:25
Targeting Molecules of “Life and Death” for Cancer Therapy
Wijnand Helfrich1, Edwin Bremer2, Marco de Bruyn1, Harald Wajant2
1Surgery, University Medical Center Groningen, Surgical Research Laboratories, 9713 GZ Groningen, The Netherlands; 2Department of Molecular Internal Medicine, Medicinal Clinic and Polyclinic II, University of Würzburg, 97070 Würzburg, Germany
Antibody-based therapeutic approaches are yielding more and more of the promise they have held since the conception of the ‘magic bullet’ theory by Paul Ehrlich. The beneficial effect of antibody-based therapies is directly related to antibody-dependent functions, such as neutralization and ADCC, but in many cases also relies on the delivery of toxic compounds to cancerous cells. However, the clinical utility of toxic antibody conjugates can be significantly hampered by side effects. Ideal effector compounds are inactive ‘en route’, but gain full activity once the antibody conjugate has bound to cancerous cells. Of significant potential in this respect are the pro-apoptotic ligands tumor necrosis factor (TNF), FasL and TRAIL. TNF ligands are normally present as homotrimeric transmembrane proteins, but can also be processed into a soluble trimeric form. Compared to their corresponding transmembrane counterpart, soluble TNF, FasL and TRAIL have a strongly reduced capacity to activate TNFR2, Fas and TRAILR2. However, all sequence information required for full activation of these receptors is latently retained in these soluble ligands and can be unmasked by oligomerization or cell surface immobilization. The latter provides a clear rationale for the use of these ligands as effectors in antibody-based therapy. The antibody-targeted ligand will be in a relatively inactive soluble form while en route. However, once bound to the targeted cancer cell the soluble TNF ligand fusion proteins will be converted into fully active membrane ligand-like molecules. Here we will, after briefly detailing the biology of TNF, TRAIL and FasL, focus on the promises and pitfalls of targeted TNF ligand fusion proteins in achieving a ‘magic bullet’ with maximum cancer selective activity and minimal side-effects.
POSTER and SHORT COMMUNICATION: Thursday 02 April, 12:50
In Vivo Biodistribution and Microvascular Binding of a High-Affinity Monoclonal Antibody Fragment F8-SIP against the Extra-domain A of Fibronectin
Güliz Parmaksiz1, Marcus Czabanka1, Dario Neri2, Peter Vajkoczy1
1Department of Neurosurgery, Charité — Universitätsmedizin Berlin, 13353 Berlin, Germany; 2Department of Chemistry and Applied Biosciences, Institute of Pharmaceutical Sciences, ETH , 8093 Zurich, Switzerland
Objective: The human monoclonal antibody fragment F8-SIP (small immunoprotein) is specific to the angiogenic marker extra-domain A of fibronectin. The aim of our study was to characterize microvascular binding and biodistribution characteristics of F8-SIP.
Methods: SF126 cells were implanted into dorsal skin chamber (DSC) preparations of nude mice. Microvascular and interstitial accumulation of F8-SIP and microvascular blood flow rate were analyzed at t = 0 h, t = 2 h, t = 4 h and t = 24 h after intraarterial application of fluorescently labeled F8-SIP (n = 5 per group) by intravital microscopy. Host vasculature of mice without tumor bearing DSCs were used as control group.
Results: F8-SIP binds specifically to tumor vessels reaching its maximum binding capacity 4 hours after injection (t = 0 h: 26.12 ± 10.89 vs. t = 4 h: 91.46 ± 6.19; p < 0.05). In control vasculature no binding was observed. Extravasation of F8-SIP to tumor interstitium was observed reaching its maximum 4 hours after injection (t = 0 h: 19.25 ± 9.45 vs. t = 4 h: 60.41 ± 7.77; p < 0.05). Microvascular binding was flow-dependent with significantly increased binding in high flow blood vessels (HF ≥ 60 nl/sec) compared to low flow blood vessels (LF ≤ 20 nl/sec) at t = 2 h by 44% and at t = 4 h by 22%, respectively. F8-SIP binding occurred preferentially in angiogenic sprouts (AS) compared to remaining tumor vasculature (RTV) 2 hours after injection (AS: 115.14 ± 12.38 vs. RTV: 77.86 ± 11.25; p < 0.05).
Conclusions: F8-SIP represents a useful tool to specifically target tumor microvessels. Microvascular binding occurs in a time- and blood flow dependent manner with preferential binding sites. Our results provide biodistribution characteristics that might be used for future clinical applications.
T-02
POSTER
Investigation of Properties of Anti-Prostate Specific Membrane Antigen-targeted Gold Nanoparticles as Drug Carriers in Tumor Therapy
Giulio Fracasso1, Vincenzo Amendola2, Cristina Anselmi1, Gabriele Marcolongo2, Sara Cingarlini3, Giorgia Cremonese1, Mariangela Figini4, Anita Boscaini1, Moreno Meneghetti2, Marco Colombatti1
1Department of Pathology — Section of Immunology, Policlinico G.B. Rossi, University of Verona, 37134 Verona, Italy; 2 Dept. Chemical Sciences, University of Padua, 35100 Padua, Italy; 3Medical Oncology Unit, University of Verona, 37100 Verona, Italy; 4Dept. of Experimental Oncology, Istituto Nazionale Tumori, 20100 Milan, Italy
Specific targeted delivery and control drug release are desirable properties of a drug for tumor therapy. Nanomedicine, the science that studies the application of nanotechnology to disease treatment, might be of help. Targeted nanoparticles (NP) for their size and structure are able to enhance the accumulation in the tumor of encapsulated or linked / adsorbed molecules (gene, drug). We have therefore investigated the binding properties of gold-NPs (20 nm) conjugated to D2/B, a mAb recognizing the prostate specific membrane antigen (PSMA). PSMA for its wide distribution in prostate tumor (about 70-80% of patients are PSMA+), is an important biomarker in the management of this malignancy using targeted drugs. Gold-NPs have been synthesized by laser ablation, mixed with a reporter solution (Texas red) and treated with a thiol-PEG solution. Derivatization of PEG-COOH with EDC/Sulfo NHS has been used to covalently link the mAb to the NPs. D2/B-NP binding to LNCaP (PSMA+) cells has been assessed by cytometry. We have measured a MFI (mean fluorescence value) of 1,383 for D2/B-NP whereas the MFI of the negative control (CTRL-) was 68. The binding specificity was confirmed on PSMA– Jurkat cells (MFI of 121 and 101 for D2/B-NP and CTRL-, respectively). Binding and internalization of D2/B-NP in LNCaP cells were assayed by confocal microscopy; detection of D2/B-NP by surface-enhanced Raman scattering also revealed the selective binding of the NPs to Ag+ cells. The results of specific delivery and internalization support our idea to use mAb anti-PSMA targeted NPs to enhance the transport of toxic drugs in the tumor.
SHORT COMMUNICATION: Friday 03 April, 12:55
Mechanistic Aspects of the Synergistic Cytotoxicity between Gypsophila Saponins and Type I Ribosome-inactivating Proteins
Alexander Weng*1, Christopher Bachran*2, Hendrik Fuchs*2, Matthias Melzig*1
1Institute for Pharmacy, Freie Universität Berlin, 14195 Berlin, Germany; 2Zentralinstitut für Laboratoriumsmedizin und Pathobiochemie, Charité — Universitätsmedizin Berlin, 12200 Berlin, Germany
* These authors contributed equally to this work
Certain saponins from Gypsophila paniculata L. were shown to enhance the cytotoxicity of the type I ribosome-inactivating protein saporin and of a saporin-based chimeric toxin in cell culture experiments. Up to now it was assumed that the Gypsophila saponin-mediated enhancement of cytotoxicity of these toxins is based on a modulation of transport processes, resulting in an enhanced endocytosis of toxin molecules. Here we show that Gypsophila saponins do not influence the endo- or exocytosis of a tritiated saporin. Further experiments with bafilomycin A1, a specific inhibitor of the vacuolar ATPase, and sub-cellular fractionation studies indicate a Gypsophila saponin-mediated endosomal escape of the N-glycosidase saporin. Since saporin-based chimeric toxins are cleaved off from their tumor-specific ligand intracellularly, it is most likely that the saponin-mediated increased cytotoxicity of these targeted tumor therapeutics is also due to a Gypsophila saponin-induced endosomal escape of the enzymatic active saporin. Our results are most important for all saporin-based chimeric toxins utilized in tumor therapy since their therapeutic potential could be drastically enhanced by Gypsophila saponins.
T-04
POSTER
Yeast as a Model for Drug Target Identification in Cancer Research
Anja Nowka
OWL, Max-Planck-Institut für Molekulare Genetik, 14195 Berlin, Germany
The budding yeast Saccharomyces cerevisiae is a well-recognized model organism for the development of genomic technologies and approaches. Importantly, many cellular mechanisms controlling essential molecular processes are conserved from yeast to higher eukaryotes; for example ~50% of yeast proteins have orthologs in human. Therefore yeast has been explored extensively, e.g. for the identification of cellular signaling pathways. Consequently, these pathways are certainly the best-characterized signaling pathways of any eukaryote. Additionally, due to the widespread structural and functional similarities between yeast and mammalian signaling systems, data generated in yeast were shown to be generally applicable to humans. Yeast is also used for studying in vivo effects of drugs. In cancer research genome-wide drug screens in yeast have been exploited for the identification of target genes and for uncovering cellular mechanism and their alterations in disease state. In such genome-wide yeast studies novel genes mediating drug-resistance were identified, e.g. for 5-fluorouracil (5-FU, an analogue of the nucleic acid uracil), cisplatin (platinum-based chemotherapeutic agent) and doxorubicin (anthracyclin). Here, the results of such genome-wide screens using camptothecin, a topoisomerase I inhibitor, as well as for imatinib, a BCR-ABL kinase inhibitor, will be presented.
T-05
POSTER
Combination of Saponin and a Chimeric Toxin Induces Apoptosis
Diana Bachran1, Stefanie Schneider1, Romy Urban1, Alexander Weng2, Matthias Melzig2, Christopher Bachran1, Hendrik Fuchs1
1Zentralinstitut für Laboratoriumsmedizin und Pathobiochemie, Charité - Universitätsmedizin Berlin, 12200 Berlin, Germany; 2Institut für Pharmazie — Pharmazeutische Biologie, Freie Universität Berlin, 14195 Berlin, Germany
Introduction: Chimeric toxins (CTs) have significantly improved targeted anticancer therapies. Previously we described the drastically increased uptake of CTs into target cells by combination with saponins [1]. Here we analyzed the ability of a CT (Sap-EGF), consisting of the ribosome-inactivating protein saporin and human EGF, to induce apoptosis in combination with saponin in a tumor cell model.
Methods: For analysis of cytotoxicity and apoptosis, HER14 cells (stably EGF receptor-expressing NIH-3T3 cells) or HeLa cells were preincubated 5 min with Saponinum album (SA) before Sap-EGF was added and incubated for 48 h. Apoptosis was analyzed by histone-associated DNA fragment formation or the cleavage of poly(ADP-ribose) polymerase 1 (PARP).
Results: The combination of SA (in a non-toxic and non-permeabilizing concentration) and Sap-EGF resulted in an enhanced and synergistic cytotoxicity on the HER14 cells. At the GI50 values DNA fragmentation and PARP cleavage was clearly detectable in both cases with and without SA, however, SA lowered the concentration necessary to induce DNA fragmentation and PARP cleavage.
Conclusion: Analysis of cell death on HER14 cells pointed out that cell death induced by Sap-EGF was mediated by apoptosis and was enhanced by SA. The triggering of apoptosis in target receptor-expressing cells does, in contrast to necrosis, not lead to cell rupture. This inhibits the release of the chimeric toxin as well as other toxic substances and therefore reduces unspecific damage on other cells.
References:
[1] Heisler I, Sutherland M, Bachran C, Hebestreit P, Schnitger A, Melzig MF, Fuchs H, J Control Release 106:123-137 (2005).
T-06
POSTER
Targeted Uptake of Natural Drug Carriers by Tumor Cells
Mira Müller*, Angelika Müller*, Nadine Sternberg, Saphira John, Pei Yun Teo, Ziyad Abdallah, Radostina Georgieva, Hans Bäumler
Berlin Brandenburg Center for Regenerative Therapies (BCRT), Charité — Universitätsmedizin Berlin, 10117 Berlin, Germany
* These authors contributed equally to this work
Treatment of cancer with chemotherapy has many well-known drawbacks. Major difficulties relate to the limited accessibility of the chemotherapeutics to cancer cells requiring high doses, their intolerable toxicity, and non-specific targeting. In an attempt to improve therapy and reduce side effects, drug delivery systems have been developed that can deliver drugs directly to the site of interest. Red blood cells (RBC) represent a potential system to carry drugs to the desired site of therapeutic action and provide an extraordinary vehicle for the dissemination of drugs in the circulation, due to the ability of their membranes to be opened and resealed. They are also biocompatible, non-immunogenic, have a large capacity and long life span. The aim of this study was to conjugate RBC with monoclonal tumor-specific antibodies, Ep-CamAua1 mAb, load them with quantum dots (qd) as a model substance for a chemotherapeutic agent, and evaluate their potential use for the specific targeting of mamma carcinoma cells (T47D). Ep-CAM mAb was attached to the RBC via a biotin / avidin-bridge. Afterwards the RBC were loaded with qd and incubated with T47D cells. Evaluation was carried out by means of flow cytometry and confocal laser scanning microscopy. The modified RBC bound to the T47D cells and had been phagocytosed by them. After uptake the qd were detected within the cancer cells. Analysis suggests that RBC present a potentially successful targeted drug delivery system as direct delivery of the loaded agent to the cancer cells was achieved.
SHORT COMMUNICATION: Friday 03 April, 15:50
Tumor Targeting Using Magnetite Loaded Red Blood Cells
Radostina Georgieva1, Nadine Sternberg1, Angelika Müller1, Munish Chanana2, Dayang Wang2, Norbert Buske1, Susanne Müller3, Ulf Teichgräber4, Hans Bäumler1
1Institute of Transfusion Medicine, Research Department, Charité — Universitätsmedizin Berlin, 10117 Berlin, Germany; 2Interfaces, Max Planck-Institute of Colloids and Interfaces, 14476 Potsdam-Golm, Germany; 3Charité — Universitätsmedizin Berlin, Neuriscience Research Center, 10117 Berlin, Germany; 4Charité — Universitätsmedizin Berlin, Institute of Radiology, 10117 Berlin, Germany
Red blood cells gained increasing interest as natural carriers for drugs, enzymes and nanoparticles providing an extraordinary long living, biocompatible and non-immunogenic vehicle with a large capacity. The aim of this study was to combine a contrast agent for MRI with the possibility for targeting via an external magnetic field in one and the same microcarrier. For this we loaded erythrocytes with nanosuspensions of biocompatible, surface tuneable, luminescent, and magnetic nanoparticles. The simplicity of the cells allowed observation of the behavior of nanoparticles in confined compartments at different conditions of the environment or in the presence of external fields. We have shown that magnetite nanoparticles coated with stimuli responsive polymers can be forced to form reversible aggregates by changing the temperature. This enhances their magnetic moment and offers an opportunity to focus the carriers at a desired location. Additionally, the surface of the cells can be modified manipulating the phosphatidylserine content of the outer lipid layer and incorporating some magnetite in the membrane to target the RES in the liver. The digestion of the red blood cells in the liver will insure the release of nanoparticles and co-incorporated drugs in the liver providing a high drug concentration for the therapy of liver carcinoma.
Acknowledgments: The financial support of the European Union (EFRE-ProFIT-10134275) is acknowledged.
POSTER and SHORT COMMUNICATION: Wednesday 01 April, 12:35
Bispecific and Bifunctional Antibody Molecules for Targeted Cancer Immunotherapy
Dafne Müller1, Roland Stork1, Emmanuelle Campigna2, Bruno Robert2, Philipp Diebolder1, Kirstin Zettitz1, Katharina Frey1, Agnes Banaszek1, Miriam Rothdiener1, Roland Kontermann1
1Institut für Zellbiologie und Immunologie, Universität Stuttgart, 70569 Stuttgart, Germany; 2Institut de Recherche en Cancérologie de Montpellier-IRCM, Université Montpellier, F-34298 Montpellier, France
Recombinant bispecific antibodies have shown to be able to retarget cytotoxic T cells to tumor cells in a MHC-independent manner, triggering effector cell activation and consecutive tumor cell death. In order to improve the pharmacokinetics of bispecific single-chain diabodies (scDb) we developed and compared four strategies: (1) fusion to the long-lasting HSA, (2) fusion to a staphylococcal albumin binding domain (ABD), (3) side-directed conjugation to a branched 40 kDa polyethylene glycol (PEG) and (4) N-glycosylation. Using scDbCEACD3 as model antibody we found a strongly improved circulation time for PEGylated scDb, scDb-HSA as well as scDb-ABD, while N-glycosylation only moderately prolonged circulation time. Considering that costimulation is an essential requirement not only to initiate T cell activation, but also for the regulation of a proper T cell response, we propose the combination of bispecific scDb with selective costimulation. We generated recombinant antibody-cytokine fusion proteins, either composed of a tumor cell-specific scFv fused to the extracellular domain of human ligand 4-1BBL or a diabody fused to the extracellular region of B7.2. These recombinant fusion proteins exhibited costimulatory properties, which were found to be concentration dependent, ligand-specific and substantially constrained to antigen-mediated cell binding.
T-09
POSTER
Loaded Red Blood Cells as Natural Microcarriers for Targeted Drug Delivery
Nadine Sternberg1, 2, Angelika Müller1, Mira Müller1, Ziyad Abdallah1, Radostina Georgieva1, Hans Bäumler1, 2
1Institute of Transfusion Medicine, Research Department, Charité — Universitätsmedizin Berlin, 10117 Berlin, Germany; 2Berlin-Brandenburg Center for Regenerative Therapies, 13353 Berlin, Germany
Systemic chemotherapy is the main treatment available for disseminated malignant disease. Though it is successful to some extent, there are main drawbacks such as limited accessibility of drugs to the tumor cells requiring high doses, intolerable toxicity and non-specific targeting. The use of biodegradable carriers to deliver therapeutics throughout the body not only improves the efficacy of the loaded drug but also prolongs its systemic activity, protects it from premature inactivation and prevents an immunological response. Modification of the carrier surface allows direct targeting of specific tissues and organs. Red blood cells (RBC) were loaded via several hypotonic dilution methods with the hydrophilic model substances bovine serum albumin (BSA) and dextran (70kDa), quantum dots as well as with a nanosuspension of the hydrophobic drug Amphothericin B. The integrity of the cell membrane was affected to a certain degree due to the different loading strategies. This was quantified by the binding of annexin-V. Subsequent surface modification was performed by the model peptide Insulin. Uptake of the surface modified RBC was shown in vitro using an RBE4 cell culture. The entrapment of bioactive substances or nanoparticles into RBC and their surface modification with antibodies or signal peptides facilitates a broad spectrum of applications. The liver targeting by artificially aged RBC or their uptake after surface modification with insulin are promising examples for the potential of these natural micro-carriers.
T-10
POSTER
Establishment and Characterization of a Xenograft Model for Human Cervical Cancer
Corinna Hoffmann1, Christopher Bachran2, Jonas Stanke1, Andreas Kaufmann1, Hendrik Fuchs2, Achim Schneider1, Günter Cichon1
1Gynäkologische Tumorimmunologie, Charité — Universitätsmedizin Berlin, 12200 Berlin, Germany; 2Zentralinstitut für Laboratoriumsmedizin und Pathobiochemie, Charité — Universitätsmedizin Berlin, 12200 Berlin, Germany
In contrast to the majority of human malignant tumors the generation of cell lines from primary cervical carcinomas is very difficult and attempts for engraftment and proliferation of primary tumor tissue in immune deficient mice has not been successful in the past. The lack of appropriate xenograft models impedes the search for improved specific therapeutic agents. We demonstrate for the first time a protocol that allows reliable and efficient engraftment of human cervical cancer in Scid beige mice. About 70% of transplanted tumors exhibited potent proliferation and multiple retransplantations were possible in 40% of tumors. Furthermore the cancer tissues preserved their tumor characteristics throughout xenotransplantation and multiple passages in mice. To demonstrate the significance of this tumor model we explored the therapeutic potency of a novel immunotoxin (SA2E). SA2E is a chimeric protein that was constructed by fusing the human epidermal growth factor and the plant protein toxin saporin. Local application of SA2E provided a substantial therapeutic effect and resulted in a reduction of tumor volume up to 60%. Reliable engraftment and high reproducibility makes this newly established xenograft model an attractive test system for new therapeutic agents against cervical cancer.
T-11
POSTER
Improved Immunotoxin Efficacy by Bacterial Tumor Targeting
Alexander Klimka1, Jochen Stritzker2, Katja Sabel1, Qian Zhang3, Elke Pogge von Strandmann4, Aladar Szalay2, 3, Martin Krönke1
1Inst. Med. Mikrobiologie, University Hospital of Cologne, 50935 Cologne, Germany; 2Department of Microbiology, University of Würzburg, Biocenter Am Hubland, 97074 Würzburg, Germany; 3San Diego Science Center, Genelux Corp., San Diego, CA 92109, USA; 4Dept. of Internal Med. I, University Hospital of Cologne, 50931 Cologne, Germany
Recombinant immunotoxins (ITs), which are usually expressed in E. coli, consist of the catalytic domain of a toxin genetically fused to a binding ligand specific for a tumor-associated antigen. The IT has to be internalized after binding to the target cell in order to be effective. But, since the efficacy of an IT is also dependent on purity, serum half-life, tumor penetration, specificity and binding affinity, ITs have to be produced and applied in high dosages and can still remain non-functional in vivo or lead to unwanted, severe side effects. To circumvent the limitations of this immunotherapeutic approach we try to combine the IT properties with the well known observation that bacteria can somehow colonize tumors and metastasis by escaping the patients immune system exclusively in close vicinity of the cancer cells. We cloned IT genes downstream of a sensitive arabinose promotor into an E. coli expression vector encoding additionally for constitutive luminescence. Attenuated E. coli transformants can be injected into tumor mouse models and screened under a low light imager for exclusive proliferation close to tumors and metastasis. After systemic administration of a non-toxic concentration of arabinose, the respective IT will be secreted by the bacteria and kill the tumor cells. Aim of the study is to visualize the reduction of tumor burdens and residual tumor cells by means of our local IT expression.
T-12
POSTER
Erythrocytes as Natural Drug Carriers of 5-Fluorouracil: Loading Procedure and Cytotoxic Efficacy
Angelika Müller1, Mira Müller1, Nadine Sternberg1, Ziyad Abdallah1, Saphira John1, Radostina Georgieva1, Bernd Paulke2, Hans Bäumler1
1Institute of Transfusion Medicine, Research Department, Charité — Universitätsmedizin Berlin, 10117 Berlin, Germany; 2Fraunhofer Institute for Applied Polymer Research, 14476 Potsdam, Germany
5-Fluorouracil (5-FU) is a widely used chemotherapeutic agent in the treatment of metastatic colorectal cancer, but its systemic application often leads to severe side effects. The reduction of these toxic effects is an important goal and achievable by specific targeting of the substance. Red blood cells (RBC) can be used as a natural drug delivery system. They can be loaded with a variety of chemotherapeutic agents by the hypo-osmotic dilution method. Depending on the procedure of surface modification it is possible to target them to different tissues. 5-FU is a very small molecule. It interacts with the RBC membrane and usually escapes from the RBC shortly after loading. We enlarged 5-FU by linking it to Albumin. Therefore 5-FU had to be changed to FU-acetate (FUAC). After activation of FUAC, it had been bound to primary amino groups of Albumin. FUAC-Albumin (FU-Alb) remained inside the RBC after loading. To compare the cytotoxic effect of FU-Alb loaded RBC with the cytotoxic effect of 5-FU and FU-Alb respectively, these agents had been incubated with colorectal cancer cells of the rat (CC531). After 48, 72 and 96 hours incubation time, the cytotoxic effect was measured by means of MTT assay. 5-FU and FU-Alb showed a similar cytotoxic effect on all breakpoints. FU-Alb RBC developed the same toxic effect, but therefore needed a longer incubation time. Additionally confocal laser scanning microscopy showed the uptake of FU-Alb RBC by the cancer cells, which induced the death of the cancer cells.
T-13
POSTER
The Genome Maintenance and Transcriptional Regulation Mechanisms of Human High Risk Papillomaviruses as Target Points for Antiviral Treatment and Cervical Cancer Prevention
Andrea Wunderlich, Michal-Ruth Schweiger
Prof. Dr. H. Lehrach, Max Planck Institute for Molecular Genetics, 14195 Berlin, Germany
Cervical cancer is the second most common cancer among women worldwide. Every year approximately 500,000 new cases are diagnosed and 270,000 women die from this malignancy. The tumor is caused by a persistent infection with high-risk human papillomaviruses (HPV). The recently developed vaccination prevents infections with high-risk HPV 16 and HPV 18, two of the most frequent virus types. Nevertheless, there are several unsolved problems: The vaccination does not cover all high risk HPV types, there is no effective treatment for infected women available and the vaccine is still not sufficiently available in the most severely affected regions, in developing countries. Therefore, an antiviral therapy would be highly desirable. During papillomaviral infections the viral genomes are tethered to mitotic chromosomes to pass the infection on to the next generation of cells during replication. For this maintenance mechanism the viral E2 protein binds to the cellular bromodomain protein Brd4. Our previous work has also established that the Brd4-E2 interaction is involved in viral transcriptional regulation mechanisms, which are involved in cervical cancer pathogenesis. The goal of the presented work is to analyze and further characterize the protein-protein interaction networks underlying these genome maintenance mechanisms and thereby to gain additional insights into the mitotic complexes involved. Furthermore, we are working on the development of target-specific anti-HPV drugs and will discuss the impact for the prevention of cervical cancer.
T-14
POSTER
Synthesis of Spacer-linked Bivalent Estrogen Analogs
Min Shan1, Anja Wellner2, Ronald Gust2, Rainer Haag1
1Institut für Chemie, Freie Universität Berlin, 14195 Berlin, Germany; 2Institut für Pharmazie, Freie Universität Berlin, 14195 Berlin, Germany
Estrogen receptor (ER) has been reported to exist as a dimer even in the absence of ligand. In order to stabilize the dimerization of ER, two hormonally active compounds with a long and exactly defined spacer are synthesized, which could interact with the ligand binding domain of ER α or ER β. Thus, the ER-intracellular signaling cascades, the cofactor recruitment and the concentration of receptor could be influenced. The effective length of a spacer for the optimum bivalent ligand is 3.9 nm, as estimated by the crystal structure of the dimeric ER [1]. We report the novel synthesis of bivalent estrogen analogs base on endoxifen and the polyethylene glycol (PEG) chains with rigid units (i.e. 10 PEG units correspond to about 4 nm long), so that we could reduce possible degrees of freedom of the bivalent estrogen analogs [2-5]. The bivalent estrogen analog was investigated for the cytotoxicity and the (anti‑)estrogenic effect. The bivalent estrogen analog showed the expected antiestrogenic effect with an IC50 of 1.4 x 10–10 M.
References:
[1] Source: Cambridge Crystallographic Data Base.
[2] Gust R, J Med Chem 46:1484-1491 (2003).
[3] Shena G, Org Biomol Chem 4:2082-2087 (2006).
[4] Forman BM, J Org Chem 68:9489-9491 (2003).
[5] Whitesides GM, J Am Chem Soc 129:1312-1320 (2007).
POSTER and SHORT COMMUNICATION: Thursday 02 April, 13:05
Development of Macromolecular Prodrugs Derivated from Dendritic Polyglycerol
Marcelo Calderon1, Ralph Gräser2, Felix Kratz3, Rainer Haag1
1Biologie, Chemie, Pharmazie, Freie Universität Berlin, 14195 Berlin, Germany; 2Proquinase GmbH, 79106 Freiburg I.Br., Germany; 3Tumor Biology Center, 79106 Freiburg I.Br., Germany
Utilizing the Enhanced Permeation Retention Effect (EPR) for passive targeting is one of the novel approaches for tumor-selective drug delivery. Polymer-based nanocarriers, especially those of dendritic architecture, have gained increasing interest for encapsulating or coupling diagnostic or therapeutic agents [1]. Here we report on the synthesis and characterization of conjugates of dendritic polyglycerol [2,3] with the (6-maleimidocaproyl)hydrazone derivative of doxorubicin (DOXO-EMCH) [4] using three coupling strategies. Dendritic polyglycerol was chosen for its non-immunogenicity and water-solubility and because the linear monohydroxy and terminal dihydroxy functionalities allow for structural flexibility through end group modification. The conjugates showed rapid release of doxorubicin under acidic conditions, but only marginal release at pH 7.4. The antiproliferative activity assessed against two human tumor cell lines AsPC1 LN (pancreatic carcinoma) and MDA-MB-231 LN (mamma carcinoma) showed less (2- to 10-fold) cytotoxicity than the corresponding free drugs. With respect to anti-tumor efficacy, the conjugates manifested excellent anti-tumor effects with complete tumor remissions up to day 30. No significant changes in body weight or mortality were observed even after administration of 3-fold the maximal tolerated dose for free doxorubicin.
References:
[1] Haag R, Kratz F, Angew Chem Int Ed, 45:1198-1215 (2006).
[2] Haag R. et al. J Am Chem Soc, 122:2954-2955 (2000).
[3] Frey H. et al. Rev Mol Biotech, 90:257-267 (2002).
[4] Kratz F. Expert Opin. Investig. Drugs, 16:855-866 (2007).
SHORT COMMUNICATION: Friday 03 April, 15:35
Self-Adjuvanting Messenger RNA-based Vaccines as a Novel Concept in Cancer Immunotherapy
Jochen Probst
CureVac GmbH, 72076 Tübingen, Germany
at the request of the author this abstract is only available in the printed version of the abstract book
T-17
POSTER
Monitoring of Hormone and Antibody-based Cancer Therapies by Concomitant Quantification of Circulating Epithelial Tumor Cells
Torsten Kroll1, Katharina Pachmann1, Annika Kohlhase1, Oumar Camara2, Ingo Runnebaum2, Klaus Höffken1
1Klinik für Innere Medizin II, Universitätsklinikum Jena, 07740 Jena, Germany; 2Frauenklinik, Universitätsklinikum Jena, 07740 Jena, Germany
We have established since several years a method of monitoring cancer therapy courses by quantification of circulating epithelial tumor cells (CETC) using fluorescence scanning cytometers [1]. As an example we will present data from an ongoing project of monitoring hormone therapy and our concomitant analysis of such cells. For this purpose peripheral blood was drawn from 178 breast cancer patients during hormone therapy at each visit with informed consent. These samples were treated with fluorochrome-labeled antibody against the surface human epithelial antigen (HEA, EpCAM). Antibody-labeled cells were detected with a fluorescence scanning cytometer and quantified. Our presented data provide evidence that repeated quantification of CETC during the course of therapy is suited to monitor the response of breast cancer patients to treatment during hormone therapy. This change in the number of CETC (increase or decrease) is correlated to the therapy's outcome. A repeated increase or an increase of more than tenfold in CETC was a strong indicator of looming relapse (p = 0.0001) and was predictive also for the response to subsequent aromatase inhibition therapy. Thus this method could be used in the future to guide therapy and to change in good time to other treatment options in this case e.g. aromatase inhibitors if the monitored therapy shows no apparent success.
References:
[1] Pachmann K, Camara O, Kavallaris A, Krauspe S, Malarski N, Gajda M, Kroll T, et al. J Clin Oncol. 26:1208-1215 (2008).
POSTER and SHORT COMMUNICATION: Friday 03 April, 12:35
The Combination of Immunotoxins and the Immunosuppressor Cyclosporin Results in Synergistic Anti-Cancer Effects in Vitro and in Vivo
Yvonne Andersson, Olav Engebraaten, Øystein Fodstad
Tumor Biology, Cancer Research, N-0310 Oslo, Norway
at the request of the authors this abstract is only available in the printed version of the abstract book
T-19
POSTER
New CD123- and CD96-directed scFv-Fragments for the Generation of Antibody Derivatives to Eliminate Acute Myeloid Leukemia Stem Cells
Christoph Stein*1, Michael Schwenkert*1, Markus Kügler1, Kristin Mentz1, Nina Reiff1, Christian Kellner2, Bernhard Stockmeyer3, Georg Fey1
1Chair of Genetics, University of Erlangen, 91058 Erlangen, Germany; 2Division of Stem Cell Transplantation and Immunotherapy, University Medical Center, Christian-Albrechts University of Kiel, 24105 Kiel, Germany; 3Department of Medicine 5, Division of Hematology/Oncology, University Hospital Erlangen, 91054 Erlangen, Germany
* These authors contributed equally to this work
Leukemia stem cells (LSCs) are well characterized for AML. Current therapeutic protocols focus on reducing the tumor mass but often do not reach the uniquely resistant LSCs. To achieve long-lasting therapeutic effects, it is necessary to eliminate the LSCs. Therefore, a set of surface antigens present on AML-LSCs has recently been characterized including CD123 and CD96. To achieve preferential targeting of AML-LSCs, single-chain antibody-fragments (scFvs) specific for CD123 were generated and the production of CD96-specific scFvs is in progress. Fusion proteins containing the extracellular domains of CD96 or CD123, respectively, were produced. Combinatorial phage display libraries were generated from spleen RNA of the immunized mice and panned with antigen-positive cells. ScFvs were subcloned from specifically binding phages. The scFv with highest affinity was fused to two different cell death inducing proteins, respectively. An immunotoxin generated by fusion to a truncated Pseudomonas Exotoxin A induced apoptosis of AML-derived MOLM-13 cells at nanomolar concentrations. Another fusion to a second scFv specific for the Fcγ-receptor III (CD16) generated a bispecific single chain Fv (bsscFv). This bsscFv mediated potent lysis of MOLM-13 cells in ADCC reactions at picomolar concentrations (EC50 = 211 pM), using mononuclear cells from healthy donors as effector cells. In ongoing research the new CD123-specific scFvs are incorporated into a variety of formats designed for preferential targeting of AML LSCs. Corresponding experiments are underway for CD96-specific scFvs.
T-20
POSTER
Development of an anti-CD33 Single-chain Antibody for Targeted Radionuclide Therapy of Acute Myeloid Leukemia
Peter Nicholls1, Louise Emberson1, Philip Blower2, Daniel Lloyd1
1Department of Biosciences, University of Kent, CT2 7NJ Canterbury, United Kingdom; 2Division of Imaging Sciences, Rayne Institute, St Thomas’ Hospital, King’s College London, SE1 7EH London, United Kingdom
For a significant number of acute myeloid leukemia (AML) patients — including older patients (> 55 years) who are not suitable for transplantation, those with high-risk cytogenetics, and those with tumors refractory to chemotherapy — conventional treatment often does not lead to a favorable outcome. These groups of patients would benefit from an alternative targeted therapy, and to this end we are developing a radiolabeled anti-CD33 single-chain antibody (scFv) to specifically ablate AML cells via the CD33 antigen. To investigate the potential of this reagent for imaging and therapy of AML, conjugation to radioisotopes such as 99mTc, 188Re, and 111In is being undertaken, and radiobiological properties and biodistribution/dosimetry evaluated. The Pichia pastoris expression system has been used for the production of the anti-CD33 scFv, routinely yielding 120-140 mg/l (complex media) and 15-30 mg/l (minimal media), which is purified using an Ni2+ HiTrap column. Flow cytometry and confocal microscopy have demonstrated that the anti-CD33-scFv binds specifically to CD33+ HL60 and U937 cells, and is internalized at a rate comparable to that of the parent monoclonal antibody. Radiolabeling of the scFv with 99mTc via the His-tag was performed using the Mallinckrodt IsolinkTM reagent. Labeling was evaluated using ITLC and phosphorimaging, and HL60 cell binding studies demonstrated that significantly more radiation was associated with cells incubated with 99mTc-anti-CD33-scFv than with 99mTc alone. Further in vitro evaluation of the labeled reagent is currently in progress.
Category: Targeted Tumor Therapies in Clinical Applications
Luzie Fabisch Memorial Lecture
TALK: Wednesday 01 April 2009, 11:15
Anti-CD3 Recombinant Diphtheria Immunotoxin Therapy of Cutaneous T Cell Lymphoma
Arthur Frankel1, Sandy Zuckero2, Allison Mankin2, Margarite Grable2, Kindra Mitchell2, Yu-Jen Lee2, David Neville3, Jung Woo2
1Internal Medicine, Executive Director, Cancer Research Institute of Scott & White, Hematology / Oncology, Temple, TX 76502, USA; 2Hematology / Oncology, Internal Medicine, Cancer Research Institute of Scott & White, Temple, TX 76502, USA; 3Angimmune LLC, Bethesda, MD 20814, USA
The recombinant CD3 immunotoxin, A-dmDT390-bisFv(UCHT1), composed of the catalytic and translocation domains of diphtheria toxin fused to two single chain Fv fragments of an anti-CD3ε monoclonal antibody, was administered to five patients with cutaneous T cell lymphoma by eight 15 min intravenous infusions over four days. Side effects were fever, chills, nausea, hypoalbuminemia, transaminasemia and reactivation of Epstein-Barr virus and cytomegalovirus. Half-life of drug was 40 min. Anti-immunotoxin antibodies developed in all patients after two weeks. Two patients had partial remissions lasting 1 and 6+ months. The agent is undergoing further dose escalation and shows promising results in this disease.
TALK: Friday 03 April 2009, 12:00
NK Cell-based Immunotherapy of Malignant Diseases
Achim Rothe1, Ron Jachimowicz1, Katrin Reiners1, Jörg Keßler1, Peter Borchmann1, Giulio Fracasso2, Marco Colombatti2, Paul J Yazaki3, Barbara E. Power4, Andreas Engert1, Elke Pogge von Strandmann1
1Department I of Internal Medicine, Laboratory of Immunotherapy, University Hospital Cologne, Joseph Stelzmann Str. 9, 50931 Cologne, Germany;
2Department of Pathology — Section of Immunology, Policlinico G.B. Rossi, University of Verona, 37134 Verona, Italy; 3Division of Molecular Biology, Beckman Research Institute of the City of Hope, Department of Radioimmunotherapy, Duarte, CA, USA;
4Barbara Power Patrys Ltd, BIO21 Institute, 30 Flemington Road, Parkville 3010, Victoria, Australia
The use of immunostimulatory antibodies that engage key cell-surface receptors expressed on cytotoxic effector cells (T-cells, NK cells) is a promising strategy to augment immune responses to cancer. Such immunostimulatory mAbs potentiate immune cell responses by acting as ligands to co-stimulatory molecules, either antagonizing those that suppress immune responses or activating others that amplify immune responses. The development of bispecific constructs called “immunoligands” combine the engagement of immune receptors with the targeting of tumor specific antigens. The proof-of-principle construct is a fusion protein consisting of an antibody-derived single chain detecting CD138 (syndecan I) on multiple myeloma cancer cells and a ligand with specificity for the triggering NK cell receptor NKG2D. In vitro and in vivo mouse experiments demonstrate that this fusion protein can trigger both, NK-cell-mediated killing of tumor cells and NK cell-dependent cytokine secretion resulting in impressive anti-tumor effects. Novel immunoligands targeting antigens expressed on solid tumors such as PSMA (prostate) and CEA (colon carcinoma) that are fused to ULBP2, a ligand for NKG2D and to BAT3, a ligand for the natural cytotoxicity receptor NKp30 will be presented. Since the efficacy of immunoligands depends on a simultaneous binding to tumor cells and immune effector cells we expect that the in vivo activity should be restricted to the tumor microenvironment limiting the risk for the therapy-induced autoimmune reactions or chronic inflammation.
TALK: Friday 03 April 2009, 15:00
ED-B Fibronectin: Neoangiogenesis and Drug Targeting
Horst Dürkop1, Stefanie Sauer1, Paola Erba2, Mario Petrini3, Andreas Menrad4, Leonardo Giovannoni5, Chiara Grana6, Burkhard Hirsch1, Luciano Zardi7, Giovanni Paganelli6, Giuliano Mariani2, Dario Neri8, Hans D. Menssen9
1Institut für Pathologie, Charité — Universitätsmedizin Berlin, CBF, 12200 Berlin, Germany; 2Regional Center of Nuclear Medicine, University of Pisa Medical School, Pisa, Italy; 3Department of Hematology, University of Pisa Medical School, Pisa, Italy; 4Genzyme Europe Research, Cambridge Science Park, Cambridge, United Kingdom; 5Philogen, SpA, Siena, Italy; 6Department of Nuclear Medicine, European Institute of Oncology, Milan, Italy; 7Laboratory of Innovative Therapies, Centro Biotechnologie Avanzate, Genova, Italy; 8Department of Chemistry and Applied Biosciences , Institute of Pharmaceutical Sciences, Swiss Federal Institute of Technology, ETH Zurich, Switzerland; 9Department of Global Clinical Development Oncology, Bayer Health Care Pharma, Montville, NJ, USA
Current treatment of malignancies often involves large doses of rather unspecific drugs, frequently resulting in severe adverse events. Thus, modern clinical cancer research focuses on compounds able to discriminate between malignant and normal tissues. The extra-domain B of fibronectin (ED-B FN) is extracellularly expressed around newly formed blood vessels. It is highly expressed in cancers but not in normal mature tissues. Therefore, it is a promising target for selective cancer therapies. We established a novel immunohistologic epitope retrieval technique for paraffin-embedded tissues, and were able to analyze ED-B FN expression in biopsies of more than 300 patients with Hodgkin, Non-Hodgkin lymphoma, and myeloproliferative neoplasms. ED-B FN expression was nearly absent in normal lymph nodes and bone marrow biopsies. The extent of vascular ED-B FN expression in lymphoma tissues was positively correlated with grade of malignancy. ED-B FN expression was enhanced in lymph nodes with severe lymphadenopathy and in some hyperplastic tonsils. The in vivo accessibility of ED-B FN was confirmed in three lymphoma patients, in whom the lymphoma lesions were visualized on scintigraphy with 131I-labeled L19 small immunoprotein (131I-L19SIP). In two relapsed Hodgkin lymphoma patients, 131I-L19SIP radioimmunotherapy induced a sustained partial response, qualifying ED-B FN as a promising target for antibody-based lymphoma therapies.
C-01
POSTER
BRCA1-mutation Status and Polymorphisms Significantly Influence the Histological Grading and Lymph Nodes Involvement in Ovarian Cancer Patients
Desislava Dimitrova1, 2, Elena Ioana Braicu1, 2, 3, Radoslav Chekerov1, 2, Gülten Oskay-Özcelik1, 2, Werner Lichtenegger1, 2, Sven Olek4, Jalid Sehouli1, 2
1Department of Gynecology, Charité Medical University, 13353 Berlin, Germany; 2Charité Medical University, European Competence Center for Ovarian Cancer, 13353 Berlin, Germany; 3First Clinic of Obstetrics and Gynecology, Medical University, Cluj-Napoca, 400000 Cluj-Napoca, Romania; 4Epiontis GmbH, 12489 Berlin, Germany
Introduction: Germ line mutations of BRCA1 and BRCA2 tumor suppressor genes are involved in 5-15.3% of all epithelial ovarian cancers and in up to 80 % of the hereditary ovarian cancers. Patients with BRCA1 mutation have generally high-grade and advanced-stage serous ovarian carcinomas. Despite these unfavorable histological features, BRCA1-associated cancers seem to have better clinical outcome than sporadic cancers. The objective of our study was to screen a population based-group of primary ovarian cancers for BRCA1 mutations and to correlate the BRCA1-mutation status with the classical clinical prognostic factors as also to analyze the impact on overall and disease free survival. Hereby we present data from our current interim analyzes.
Methods: Sixty-eight patients with primary ovarian cancer were screened for BRCA1 mutations in exon 11 of BRCA1 gene using direct sequencing. All samples were obtained from the systematic Tumor bank Ovarian Cancer (www.TOC-Network.de).
Results: The median age of the patient was 53 years (range 33-74). Most of the cases were diagnosed with advanced FIGO stage (73.5%), high grading (55.9%) and serous histological type (82.4%). Five mutations with previously reported clinical importance were found (7.4%) — 4 deletions and 1 insertion. In this study 10 (14.7%) patients were heterozygous carriers for 1186 polymorphism with unknown clinical importance. Our study showed a significant correlation (P = 0.04) between BRCA1 positive mutation status and grading. The presence of 1186AG polymorphism correlated significantly (P < 0.01) with the presence of lymph node metastasis.
Conclusions: Our study showed that BRCA1 mutations and polymorphisms are associated with poor differentiated ovarian cancer and lymph nodes involvement.
Acknowledgments: This project was kindly supported by Berliner Krebsgesellschaft e.V.