ABSTRACTS FOR POSTERS AND SHORT COMMUNICATIONS 2006
Click on ![]() left of the speaker's name in the program to display the corresponding abstract of invited speakers.
left of the speaker's name in the program to display the corresponding abstract of invited speakers.
CATEGORIES:
Tumor-associated antigens as targets for immunotoxins (A)
Targeted tumor therapies in clinical applications (C)
Molecular drug optimization (M)
Role of toxin selection (T)
Other (O)
Category: Tumor-associated antigens as targets for immunotoxins
A-01
POSTER
Ligand-Mediated Modulation of the Type III Tyrosine-Kinase Receptor c-kit is a possible specific Entrance Mechanism for Immunotoxins in c-kit+ Leukemia
Julia Sadowski1, Ulrike Erben2, Stefan Dübel3, Eckhard Thiel1, Michael Notter1
1Charité, Campus Benjamin Franklin, 12200 Berlin, Germany; 2Gastroenterology, Infectiology and Rheumatology, Charité Campus Benjamin Franklin, 12200 Berlin, Germany; 3Biochemistry and Biotechnology, Technische Universität Carolo-Wilhelmina, 38106 Braunschweig, Germany
The tyrosine-kinase receptor c-kit is present in 70-80% of acute myeloid leukemia cases. A SCF-IgG1 fusion protein containing the hinge (H)-domain was produced in COS1 and J558 cells as trimeric molecule in low concentrations (Erben et al. Cancer Res, 1999). SCF-IgG1 proved at least 3.5-fold more potent than native equimolar concentrations of SCF in modulating c-kit. We questioned whether the SCF-mediated modulation of c-kit allows to directly and specifically deliver toxic compounds into c-kit+ leukemia cells. Dimerized human SCF molecules containing the H region and various C-terminal proteins were constructed including a reporter linking SCF-H to an enhanced green fluorescent protein (EGFP). SCF-H-EGFP was predominantly released as a dimeric or oligomeric molecule by transfected eukaryotic cells. Cells of the c-kithi cell line CS-1 were incubated with SCF-H-EGFP. The EGFP signal as determined by flow cytometry diminished by 50% within 2 h and was present for at least 3 h. Human ribonuclease A (HPR) was cloned 3´ of the SCF-H gene to create an immunotoxin. Uptake of this construct into CS-1 cells studied by intracellular staining for SCF-H-HPR revealed a time course comparable to the SCF-H-EGFP signal. In conclusion, multimeric SCF ligands appear to be promising carrier molecules for targeting toxic proteins into c-kit expressing leukemic cells via ligand triggered receptor modulation.
COMPANY PRESENTATION: Thursday 30 March 2006, 16:45
![]()
Heterologous Virus-Like-Particles: Recombinant Nanosystems as Versatile Antigen Delivery Devices for Cancer Therapy
Juergen Hess, Christoph Reichel, Claus Ruehland, Christian Reiser
Immunotherapy, responsif GmbH, 91056 Erlangen, Germany
Recombinant murine polyomavirus-like-particles represent promising candidates in the development of novel immunotherapeutics against cancer. These recombinant nanoparticles consist of VP1, the major coat of polyomavirus, that spontaneously assembles into these highly ordered, icosahedral structures [1]. By means of genetic engineering these nanostructures successfully serve as carrier matrix with per se adjuvant activity for the delivery of guest epitopes, protein fragments or complete proteins. Using this nanobiotechnology, potent innate and adaptive immune responses with emphasis on antibodies and T cells are induced against self or non-self foreign antigens [2, 3]. Breaking of tolerance required for therapeutic interventions against cancer represents the hallmark of such an outstanding antigen delivery system. In combination with the increasing identification of validated target antigens from tumours, and promising progress in bioprocess development, such nanostructures offer novel treatment opportunities against cancer.
[1] Abbing et al. Efficient intracellular delivery of a protein and a low molecular weight substance via recombinant polyomavirus-like particles. J. Biol. Chem. 279: 27410-27421 (2004).
[2] Velupillai et al. Polyomavirus-like-particles elicit polarized cytokine responses in APCs from tumor-susceptible and resistant mice. J. Immunol. 176: 1148 (2006).
[3] Tegerstedt et al. A single vaccination with polyomavirus VP1/VP2Her2 virus-like-particles prevents outgrowth of HER-2/neu-expressing tumours. Cancer Res. 65: 5953 (2005).
A-03
A CD33-specific Single-Chain Immunotoxin Mediates Potent Apoptosis of Cultured Human Myeloid Leukemia Cells
Michael Schwemmlein1, Matthias Peipp2, Karin Barbin1, Domenica Saul1, Bernhard Stockmeyer3, Roland Repp4, Josef Birkmann5, Fuat Oduncu6, Bertold Emmerich6, Georg Fey1
1Chair of Genetics, University of Erlangen-Nuremberg, 91058 Erlangen, Germany; 2Department of Nephrology and Hypertension, University Clinic, University of Schleswig-Holstein, 24105 Kiel, Germany; 3Department of Medicine III, University of Erlangen-Nuremberg, 91054 Erlangen, Germany; 4Section of Stemcell- and Immunotherapy, University Clinic, University of Schleswig-Holstein, 24105 Kiel, Gemany; 5City Hospital, Nuremberg, Medical Clinic 5, 90419 Nuremberg, Germany; 6Medical Clinic Innenstadt, University Clinic Munich, 80337 Munich, Germany
CD33 is a well-suited target for treatment of acute myeloid leukemia (AML) with antibodies or derived agents, because the leukemic blasts of more than 90% of AML patients express this antigen. CD33 is also expressed on a subset of infant acute lymphoblastic leukemias (ALL), whereas hematopoietic stem cells, lymphoid cells, and non-hematopoietic cells lack CD33. An IT was constructed from a newly isolated CD33-specific single chain Fv (scFv) antibody fragment by combining the scFv with an engineered variant of Pseudomonas Exotoxin A (ETA). This so-called CD33-ETA IT carries the KDEL peptide at its C-terminus, a cellular peptide mediating improved retrograde transport to the endoplasmic reticulum (ER). The purified recombinant fusion protein induced apoptosis of the human myeloid cell lines U937, HL-60 and THP-1. Up to 98% of U937 cells were eliminated after treatment for 72h with a single dose of 500 ng/ml (~ 7 nM). Killing was antigen-specific and occured by apoptosis. In a direct comparison, CD33-ETA showed no cytotoxic activity on antigen-negative T-ALL-derived CEM cells, whereas the clinically approved agent Gemtuzumab Ozogamicin (Mylotarg™), a monoclonal anti-CD33 antibody coupled to the cytotoxic compound calicheamicin, significantly lysed antigen-negative CEM cells. CD33-ETA also mediated apoptosis of fresh patient-derived AML cells from bone marrow and peripheral blood. The pronounced antigen-restricted cytotoxicity of the new IT and its action in low nanomolar concentrations makes it a candidate for further evaluation of its therapeutic potential.
Tumor-Associated CD75s-Gangliosides and CD75s-Bearing Glycoproteins with Neu5Acα2-6Galβ1-4GlcNAc-Residues are Receptors for the Anticancer Drug rViscumin
Iris Meisen1, 2, Bernhard Kniep3, Jörg Haier4, Norbert Senninger4, Ulrich Neumann5, Martin Langer6, Klaus Witthohn7, Jadranka Milosević1, Jasna Peter-Katalinić1, Johannes Müthing1
1Institute for Medical Physics and Biophysics, University of Münster, 48149 Münster, Germany, 2Institute of Occupational Medicine, University of Münster, 48149 Münster, Germany, 3Institute for Immunology, University of Dresden, 01307 Dresden, Germany, 4Department of General Surgery, Münster University Hospital, 48149 Münster, Germany, 5Clinic of Poultry of the Hannover School of Veterinary Medicine, 30559 Hannover, Germany, 6BRAIN AG, 64673 Zwingenberg, Germany, 7Cytavis Biopharma GmbH, 22525 Hamburg, Germany
The anticancer drug rViscumin, currently under clinical development, has been shown in previous studies to be a sialic acid specific ribosome inactivating protein (RIP). Comparative binding assays with the CD75s-specific monoclonal antibodies HB6 and J3-89 revealed rViscumin to be a CD75s-specific RIP due to identical binding characteristics toward CD75s-gangliosides. The receptor gangliosides are IV6nLc4Cer, VI6nLc6Cer, and the newly characterized ganglioside VIII6nLc8Cer, all three carrying the Neu5Acα2-6Galβ1-4GlcNAc-motif. In order to elucidate the clinical potential of the rViscumin targets, CD75s-gangliosides were determined in several randomly collected gastrointestinal tumors. The majority of the tumors showed an enhanced expression of CD75s-gangliosides compared to the unaffected tissues. The rViscumin binding specificity was further investigated with reference glycoproteins carrying sialylated and desialylated type II N-glycans. Comparative Western blots of rViscumin and ricin, an rViscumin homologous but galactoside-specific RIP, revealed specific recognition of type II N-glycans with CD75s-determinants by rViscumin, whereas ricin failed to react with terminally sialylated oligosaccharides such as CD75s-motifs and others.
This strict binding specificity of rViscumin and the increased expression of CD75s-gangliosides in various tumors suggest this anticancer drug as a promising candidate for an individualised adjuvant therapy of human tumors.
Synergistic Anticancer Effects In Vitro and In Vivo of Pseudomonas Exotoxin-containing Immunotoxins and a Non-cytostatic Drug in Clinical Use.
Yvonne Andersson, Olav Engebraaten, Øystein Fodstad
Tumor Biology, Cancer Research, N-0310 Oslo, Norway
Immunotoxins belong to a group of targeted therapy candidates for being tested in patients where standard cancer treatment is no longer an option. Two of our ITs containing Pseudomonas exotoxin A, MOC31PE (anti-EpCam/EGP-2)and BM7PE (anti-mucin1), have shown promising preclinical effects and MOC31PE is currently in a phase I/II clinical trial. Further improvement in the immunotoxin field, however, is clearly dependent on increased knowledge of the molecular mechanisms by which the ITs affect the cancer cells.
We have recently found that our ITs induced several parallel signalling events. Initially an early activation of mitochondrial-dependent survival pathways to protect the cells against the toxic effects of the immunotoxin was observed. Subsequently, the immunotoxin-induced cytotoxic effects efficiently killed the cancer cells by two mechanisms, inhibition of protein synthesis and induction of apoptosis.
The obtained knowledge has been used with the objective to further improve immunotoxin efficacy, and we have found that a combination of MOC31PE or BM7PE and a drug commonly used for other clinical conditions than cancer, was investigated in vitro and in animal models. The combination caused a remarkable reduction in ID50 of each IT and a synergistic effect on IT-induced cell death. Furthermore, the combination therapy was shown to strongly inhibit formation of metastases in nude rat models for breast or cervical cancer. In conclusion, the combination might represent an effective anti-metastatic therapy without the side effects often observed with IT therapy.
A-06
Exposure of Melanoma Cells to 9.2.27PE, an Immunotoxin Targeting the HMW-MAA Antigen, causes Apoptosis via the Caspase Dependent Pathway
Karianne Risberg, Øystein Fodstad, Yvonne Andersson
Dept. of Tumor Biology, The Norwegian Radium Hospital, 0383 Oslo, Norway
The incidence of melanoma is increasing in Norway and in many other countries. Dacarbazine is still considered the standard drug in chemotherapy of melanoma, although the response rate is at maximum 20% and the complete response rate is less than 5%. Hence, there is an obvious need for new therapies, preferably approaches that have different mechanisms of action.
We have used an immunotoxin consisting of the 9.2.27 antibody covalently linked to the Pseudomonas Exotoxin A. The 9.2.27 antibody binds with high affinity and specificity to the HMW-MAA antigen expressed on most malignant melanomas and melanoma cell lines. The 9.2.27PE induced inhibition of protein synthesis and apoptosis in FEMX malignant melanoma cells. Western Blot analysis showed that 9.2.27PE induced activation of caspase-3 and PARP inactivation in the FEMX cells, suggesting that apoptosis is induced via the caspase-dependent pathway. In addition, a rapid decrease of Livin/ML-IAP and the anti-apoptotic protein Mcl-1 and were observed, whereas the levels of the anti-apoptotic proteins Bcl-2, Bcl-xl and Survivin remained unchanged. It has been suggested that Livin, an inhibitor of apoptosis, might play an important negative role in the treatment of melanoma as Livin overexpression causes drug resistance in vitro. Interestingly, we have preliminary results indicating that 9.2.27PE causes decreased cell viability in Dacarbazine resistant melanoma cells. In summary, 9.2.27PE causes cell death through a caspase dependent pathway and is a potential candidate drug for treatment of drug resistant malignant melanomas.
A-07
![]()
Expression of ED-B fibronectin in human malignant lymphoma
S. Sauer1, Andreas Menrad2, Horst Dürkop1, H. D. Menssen2
1Institut für Pathologie, Charité – Universitätsmedizin Berlin, Campus Benjamin Franklin, 12200 Berlin, Germany; 2Corporate Research Oncology, SCHERING AG, 13342 Berlin, Germany
Angiogenesis is crucial for cancer growth. Newly formed blood vessels of solid cancer tissues are strinkingly different from normal healthy counterparts. They are characterized by vascular shunts, uneven vessel diameters, endothelial fenestrations, and contain an extra-domain B of fibronectin (ED-B). Since not detectable in normal adult tissues, ED-B seems to be well suited for vascular targeting. For hematological malignancies, ED-B expression has not been studied systematically. Human samples of different hematological malignancies, non-malignant or reactive lymphatic conditions, and normal lymphatic and hematopoietic tissues were analyzed, using immunohistochemistry (IHC). IHC for ED-B detection was only possible on cryosections until recently. Steam pressure-based recovery was used on paraffin-embedded tissue sections to detect different ED-B epitopes by IHC with the mAbs BC1, L19, and the newly created MX1. No ED-B was detectable in normal lymph nodes and lymphatic tissues. But in almost all lymphoma entities vessel-associated ED-B was found to a substantial but varying extent in the following number of cases: Hodgkin lymphoma in 43/45, diffuse large B cell lymphoma in 17/20, follicular lymphoma in 30/31, mantle cell lymphoma in 14/15, B-CLL in 9/10, multiple myeloma in 10/10, and anaplastic large cell lymphoma in 11/12 samples. Reactive lymph nodes (9/10) showed a faint ED-B expression in the vascular walls, whereas in hypertrophic tonsils and healthy bone marrow ED-B was only found very rarely. Our results indicate for the first time the presence of vessel- and stroma-associated ED-B in almost all malignant lymphoma entities, laying the foundation for ED-B based vascular immunotargeting in lymphoma treatment.
![]()
Therapeutic efficacy of the targeted immunocytokine L19-IL2 in orthotopic animal models for pancreatic- and hepatocellular carcinoma
Andreas Menrad1, K. Wagner2, L. Zardi3, D. Neri3, K. Bosslet1
1Corporate Research Oncology, SCHERING AG, 13342 Berlin, Germany; 2Charité - Universitätsmedizin Berlin, 10117 Berlin, Germany, 3PHILOGEN, 53100 Siena, Italy
Introduction:
Despite being approved for the clinical treatment of metastatic renal cell carcinoma, interleukin 2 (IL2) can induce severe toxic side effects after systemic application. Local administration of IL2 has been somewhat more effective and has resulted in the control of malignant effusions and the generation of significant remissions of established lesions. Since the toxicity of IL2 is dose-limiting, the full potential of this cytokine for cancer therapy has not been fully realized. To circumvent these obstacles, an alternative approach is the use of targeted IL2-fusion proteins which can accumulate at the tumor to cause high local concentrations of the cytokine for improved anti-tumor activity. In collaboration with PHILOGEN, we have investigated the recombinant targeted immunocytokine L19-IL2 consisting of the human targeting scFv-antibody L19 and the human cytokine IL2 as the therapeutically active effector moiety. L19-IL2 binds to ED-B fibronectin which is characterized as one of the most selective oncofetal antigens associated with the process of neo-angiogenesis.
Results:
Hepatocellular carcinoma and pancreas carcinoma show a perivascular and stromal expression of ED-B fibronectin within the tumor tissue whereas normal tissue and tissue samples of pancreatitis and liver cirrhosis show no positive staining of ED-B-fibronectin. Three clinically relevant orthotopic animal models for HCC and RC have been used to establish the therapeutic potential of the targeted immunocytokine L19-IL2. We could demonstrate that 1) The ED-B-fibronectin expression in these animal models is comparable to the human situation, 2) L19-IL2 has a superior efficacy compared to non-targeted IL2: Tumor stasis was achived at clinically relevant doses of L19-IL2, whereas animals treated with non-targeted IL2 showed only minor or no signs of therapeutic efficacy, 3) minor amounts of ED-B expression are sufficient for therapeutic efficacy of the targeted immunocytokine L19-IL2.
Conclusion:
These preclinical results support the initiation of clinical studies to be performed using the targeted immunocytokine L19-IL2 in cancer indications with high medical need such as pancreatic cancer and hepatocellular carcinoma.
A-09
POSTER
A Cleavable Molecular Adapter Reduces Side Effects and Concomitantly Enhances Efficacy in Tumor Treatment by Targeted Toxins in Mice
Iring Heisler1, Christopher Bachran1, Mark Sutherland1,3, Horst Dürkop2, Hendrik Fuchs1
1Institut für Klinische Chemie und Pathobiochemie, Charité – Universitätsmedizin Berlin, Campus Benjamin Franklin, 12200 Berlin, Germany; 2Institut für Pathologie, Charité – Universitätsmedizin Berlin, Campus Benjamin Franklin, 12200 Berlin, Germany; 3Present address: Institute of Cancer Therapeutics, University of Bradford, BD7 3AY Bradford, United Kingdom
The main problems associated with the administration of receptor-targeted toxins in tumor therapy are severe systemic side effects and the low transfer of the toxins into the cytosol after binding to the tumor cell surface. To improve chimeric toxins we have developed a molecular adapter that links the toxic moiety and ligand. The adapter is designed to improve cytosolic uptake, retain the toxin inside the cytosol and detoxify the drug after cell death. The toxic moiety saporin is linked either directly or via the adapter to epidermal growth factor (EGF). To determine the toxicity of the chimeric toxin in vivo, different doses (3 µg, 10 µg or 30 µg) of the adapter-containing toxin (SA2E) were given subcutaneously to BALB/c mice. The adapter-free construct (SE) as well as the free toxin (SAP) served as control under otherwile identical conditions. The lethal dose for BALB/c mice was three times less for SA2E than for SE. Furthermore, in comparison to untreated mice, SE only reduced the average weight of induced tumors by 33% whereas SA2E-treated mice exhibited 71% reduction with an almost complete suppression in 60% of the cases. In addition, severe side effects like hyperalgesia, alopecia and death were drastically reduced in SA2E-treated animals. These results clearly show that the introduction of the adapter increases the efficacy of the active drug and also decreases side effects. Since the adapter was developed for a universal applicability as described above, the data demonstrate a promising tool for the target cell-specific treatment with other active compounds.
POSTER and SHORT COMMUNICATION: Thursday 30 March 2006, 18:15
A Tumor Protease-Activated Pore Forming Toxin as a Specific Anticancer Tool
Cristina Potrich1, Rossella Tomazzolli1, Mauro Dalla Serra1, Gregor Anderluh2, Petra Malovrh2, Peter Macek2, Gianfranco Menestrina1, Mayra Tejuca3
1Istituto di Biofisica, CNR-ITC, 38050 Povo (Trento), Italy, 2Department of Biology, Biotechnical Faculty , University of Ljubljana, 1000 Ljubljana, Slovenia, 3Departamento de Bioquimica, Facultad de Biologia, Universidad de la Habana , 23 La Habana, Cuba
Equinatoxin II is a pore forming toxin from a sea anemone able to kill very unspecifically most cell types by the membrane-perturbing action of an amphiphilic α-helix located at its N-terminal. A normally active N-terminal mutant, containing one single cysteine in this α-helix, becomes totally inactive when it is bound to avidin via a biotinylated linker. We were able to construct an enzymatically activable conjugate by choosing, as a linker, a peptide containing a tumor protease cleavage site designed in order to be digested by both cathepsin B and matrix metalloproteases (MMPs). After having measured the enzymatic activity of fibrosarcoma and breast carcinoma cells, we analysed the cytotoxic effect of the conjugate on the same cell lines and on human red blood cells (HRBC) as controls. We found that the conjugate was activated, at least in part, by the tumor cell lines used, whereas it was inactive on HRBC. Three lines of evidences indicated that the activation process was dependent on the enzymatic action of cathepsin B and MMPs: 1) binding occurred normally on all type of cells including HRBC which however were insensitive being devoid of enzymes; 2) the cytotoxic effect correlated with the amount of cathepsin B activity expressed by the cells; 3) conjugate activation was reduced by specific inhibitors of cathepsin B and MMPs. These results demonstrate the possibility of tumor cell killing by a pore-forming toxin conjugate specifically activated by tumor proteases.
Category: Targeted tumor therapies in clinical applications
Discovery of Tumor Selective Peptides for Targeted Therapy of Medullary Thyroid Carcinoma
Miriam Böckmann1, Gero Hilken2, Anke Schmidt1, Aaron Cranston3, Andrea Tannapfel4, Matthias Drosten1, Brigitte Pützer1
1Vectorology & Experimental Gene Therapy, University of Rostock, Biomedical Research Center, 18055 Rostock, Germany; 2Central Animal Laboratory , University of Essen Medical School, 45122 Essen, Germany; 3Department of Oncology, CIMR, University of Cambridge and Cancer Research UK , CB2 2XY Cambridge, United Kingdom; 4Department of Pathology, University of Leipzig, 04103 Leipzig, Germany
The identification of ligands which yield gene transfer specifically to tumor cells and their metastases is crucial for improved cancer gene therapy. Medullary thyroid carcinoma (MTC) represents an attractive target for gene therapeutic strategies needed to cope with metastases or recurrent disease. We have conducted a phage display procedure to select for peptides that specifically bind to transplanted tumor xenografts and orthotopically growing primary MTCs of RET-C634R transgenic mice. Two rounds of screening on primary tumors yielded multiple copies of a phage that displays a cyclic 7-amino-acid peptide, SRESPHP, with a 3000-fold increase in titer between round one and two. The selected phage showed a highly specific binding to the tumor after systemic administration, whereas binding to other organs was reduced up to 90%. After tail vein injection, homing to the tumor was substantially reduced in the presence of synthetic SRESPHP peptide, indicating that tumor phage interaction strictly depends on the displayed peptide. Immunohistochemical analysis of paraffin sections revealed direct binding of the SRESPHP peptide to MTC tissue. Moreover, this peptide also mediates binding and internalization to human MTC cells in vitro and in vivo, suggesting abundant expression of its cognate receptor in murine and human medullary thyroid carcinoma. Specific binding and internalization was also mediated by this peptide in the adenoviral context.
This work was supported by grant PU188/3-2/3-3 and PU188/5-1/5-2 from the Deutsche Forschungsgemeinschaft.
C-02
Detection of the hypoxia induced gene CA-IX activity in tumour cell line(s) for the application as a candidate gene in radiation cancer therapy: Human glioblastoma as model
Harun M. Said, Adrian Staab, Buelent Polat, Astrid Katzer, Michael Flentje, Dirk Vordermark
Dept. of Radiation Oncology, University of Würzburg, 97080 Würzburg, Germany
Background:
Carbonic anhydrase IX (CA-IX), a hypoxia marker regulated by HIF-1a is expressed in tumour cells due to the inducing effect of the hypoxic environment. The relation between tumour hypoxia and expression of CA-IX in human glioma cells had not been examined intensively.
Materials and methods:
Cultured human glioma cell lines were exposed to (5%–0.1%) O2, for 1 h – 24 h and 24 h with reoxygenation over 24 and 48 h. CA-IX was determined via Western blots and mRNA northern blots while HIF-1a by western blots.
Results:
A weak CA-IX protein expression was shown in a normoxic environment, in U251, U373 and GaMG, while, in U87-MG it reached the expression level of the positive control (100 µM desferrioxamin (DFO) / 24 h). Different hypoxia conditions displayed an increase in CA-IX (protein) till 24hrs and a relative stability over 48 hrs after reoxygenation occured with differences in CA-IX expression level between cell lines. Sodium iodoacetate (IAA) + 24h hypoxia minimized the CA-IX expression 90% compared to normoxia. CA-IX and HIF-1a expression were comparable in hypoxic and reoxygenation conditions. Differences in CA-IX mRNA expression levels between cell lines and O2 concentrations emerged.
Conclusion:
CA-IX expression is influenced by hypoxia due to transcriptional induction, and effects of environmental stresses (diminished levels of bicarbonate and glucose) and high CA-IX protein stability in reoxygenated cells. CA-IX is a stable marker of current and (or) previous chronic hypoxia, but is conditionally suitable for this function.
C-03
Universal Multishell Nanotransporters
Michal Radowski
Fachbereich Biologie, Chemie, Pharmazie, Freie Universität Berlin, Institut für Chemie und Biochemie, 14195 Berlin, Gemany
Core-shell structured nanotransporters have attracted much attention in recent years in the field of active substance transport, since they can pass biological barriers in different ways as low molecular chemical compounds. Additionally they can protect the encapsulated active substance [1]. A great need also exists for a molecular carrier system in order to change the polarity of dyes (including special markers) and other special chemical compounds to achieve compatibility with a matrix on a nanodisperse level.
Here we introduce a new molecular nanotransport system that is suitable for both polar and nonpolar chemical compounds. Therefore it can be used as an universal system for the encapsulation of various type of molecules [2,3]. The architecture of this new carrier type consists of two shells with different polarities arranged around a dendritic core. Linkers between the shells and the shell and the core can be either chemically stable or chemically labile. The liberation of the encapsulated molecules can be induced by external signals, for example, through pH change [4].
Bibliography:
[1] R. Haag, Angew. Chem. 2004, 116, 280-284.
[2] R. Haag, M. Radowski, 2004, German Patent application.
[3] M. Radowski, A. Shukla, H. v. Berlepsch, C. Böttcher, H. Rehage, R. Haag, 2006, Submitted.
[4] M. Krämer, J-F. Stumbé, H. Türk, S. Krause, A. Komp, L. Delineau, S. Prokohova, H. Kautz, R. Haag, Angew. Chem. 2002, 114, 4426-4431.
POSTER and SHORT COMMUNICATION: Friday 31 March 2006, 12:47
PDGFRA and KIT dysregulation and mutation in malignant peripheral nerve sheath tumors (MPNST)
Nikola Holtkamp1, Ali Fuat Okuducu1, Jana Mucha1, Anastasia Afanasieva1, Christian Hartmann1, Isis Atallah1, Lope Estevez-Schwarz2, Christian Mawrin3, Reinhard Friedrich4, Victor Mautner4, Andreas von Deimling1
1Neuropathology, Charité - Universitätsmedizin Berlin, 13353 Berlin, Germany; 2Department of Surgery and Surgical Oncology, Robert-Rössle-Hospital, Charité - Universitätsmedizin Berlin, 13125 Berlin, Germany; 3Department of Neuropathology, Otto-von-Guericke-University, 39120 Magdeburg, Germany; 4Department of Oral and Maxillofacial Surgery, University-Hospital-Eppendorf, 20246 Hamburg, Germany
Platelet-derived growth factor receptor alpha (PDGFRα) and c-Kit are receptor tyrosine kinases and among the targets of imatinib mesylate which is approved for treatment of cancers. We investigated 34 MPNST, 6 corresponding plexiform neurofibromas and 1 MPNST cell culture for mutations in PDGFRA and KIT. Both genes were amplified in some MPNST. Two MPNST carried somatic PDGFRA mutations. No point mutations were seen in KIT. PDGFRα expression was present in 75% of MPNST. Expression analysis of PDGFRα ligands in MPNST and neurofibromas revealed that PDGF-A was more widely expressed than PDGF-B. Focal c-Kit expression was detected in 7% of MPNST. Imatinib treatment of MPNST cell culture S462 exerted a growth inhibitory effect and prevented PDGF-AA induced PDGFRα phosphorylation. In summary, our data suggest that MPNST patients may benefit from treatment with imatinib. A clinical trail is currently performed.
POSTER and SHORT COMMUNICATION: Friday 31 March 2006, 12:55
Simultaneous activation of CD20- and Fas-apoptotic signaling; a novel therapeutic approach for B-cell malignancies
Edwin Bremer1, Bram ten Cate1, Douwe Samplonius1, Harald Wajant2, Wijnand Helfrich1
1Patholgy & Laboratory Medicine/Medical Biology/Tumor Immunology, University Medical Center Groningen, 9713 GZ Groningen, The Netherlands; 2Department of Molecular Internal Medicine, University of Wurzburg, 97070 Wurzburg, Germany
The anti-CD20 MAb Rituximab is a promising agent for the treatment of NHL. However, treatment is not curative and CD20-negative tumor frequently arise that escape from therapy. One important effector mechanism of Rituximab is the direct induction of apoptosis by cross-linking of CD20. Recently, CD20 cross-linking by Rituximab treatment was shown to augment Fas-apoptotic signaling. We integrated this exciting finding into a novel therapeutic approach aiming at the simultaneous activation of both CD20- and Fas-apoptotic signaling pathways. Hereto, we constructed a Rituximab-derived antibody fragment (scFvRit) and genetically fused it to human sFasL. This novel fusion protein, designated scFvRit:sFasL, potently induced apoptosis at picomolar concentrations in both B-cell lines and patient-derived tumor B-cells, but not in normal human B-cells. Compared to treatment with Rituximab combined with sFasL, the scFvRit:sFasL fusion protein was >200-fold more potent. Interestingly, CD20-signaling induced by scFvRit:sFasL is independent of caspase activation and raft formation, this in contrast to treatment with Rituximab and anti-Fas. Importantly, CD20-negative bystander tumor cells were also efficiently eliminated. The potent and CD20-restricted pro-apoptotic activity of scFvRit:sFasL may be of value for the treatment of B-cell malignancies.
C-06
POSTER
pH-responsive core-shell nanocarriers
Shangjie Xu, Ying Luo, Rainer Haag
Fachbereich Biologie, Chemie, Pharmazie, Freie Universität Berlin, Institut für Chemie und Biochemie, 14195 Berlin, Germany
Although the encapsulation and transport of guest molecules into the dendritic core-shell architectures have been studied by several research groups, relatively little is known about the active release of the encapsulated guest molecules by pH-triggered cleavage of the shell in the physiological pH range. In many cases the pH dependent release from dendritic core-shell architectures has only been achieved under severe conditions or by protonation of poly(propylene amine) dendrimers and their derivatives.
Here we reported a simple and general concept for the generation of core-shell-type architectures from readily accessible hyperbranched polymers. Several pH-sensitive nanocarriers have been prepared based on commercially available dendritic core structures (polyglycerol and poly-(ethylene imine)) by attaching pH-sensitive shells through imine bonds. In some cases the pH-responsive nanocarriers showed extremely high transport capacities, which is another important criteria for efficient drug delivery, and at the same time controled release of the guest molecules at phisiological pH range.
C-07
POSTER
A33scFv::CDy: A recombinant fusion protein of a single-chain antibody with yeast cytosine deaminase for selective prodrug activation
P. Markus Deckert1, Vania Coelho1, Ulf Petrausch1, Jens Dernedde2
1Hämatologie, Onkologie und Transfusionsmedizin, Charité - Campus Benjamin Franklin, 12200 Berlin, Germany; 2Institut für Klinische Chemie und Pathobiochemie, Charité, CBF, 12200 Berlin, Germany
Antibody-directed enzyme-prodrug therapy (ADEPT) utilises antibody–enzyme constructs for targeted enzyme delivery to tumours and subsequent localised activation of a prodrug. Its potential has been demonstrated in clinical studies.
Monoclonal antibody A33 recognises a cell-surface antigen that is expressed on 95% of colon cancers. In clinical trials, radiolabelled A33 localised specifically to colon cancer cells, where it was retained for several weeks while clearing within days from normal colon. Cytosine deaminase (CD) converts 5-fluorocytosine (5-FC) into 5-fluorouracil (5-FU) and has been empolyed in ADEPT.
Recombinant fusion constructs should overcome the problems of chemical antibody–enzyme conjugation including inhomogeneous products and large protein size.
Here, we report on a new ADEPT concept based on the A33 antigen and on the cloning, expression, and bifunctional activity of recombinant scFv–constructs with cytosine deaminase from yeast as a fusion partner. The A33scFv-CDy fusion DNA was designed and constructed from a phage-display generated A33 single chain fragment and the published sequence of yeast cytosine deaminase. After cloning expression optimization in Pichia pastoris, culture supernatant was functionally tested by inhibition of A33-GFP binding to antigen-positive cells and a katalytic enzyme assay. Finally, testing the complete ADEPT system in vitro, selective toxification of the prodrug 5-FC mediated by the fusion construct was demonstrated on A33-positive LIM1215 cells, reducing survival to that of cells exposed to equimolar amounts of 5-FU.
Category: Molecular drug optimization
POSTER and SHORT COMMUNICATION: Friday 31 March 2006, 12:25
Production of Ribosome-Inactivating Proteins and Immunotoxins in a yeast Pichia pastoris Expression System
Roberta Traini1, Erika Barison1, Teresa Lopardo2, Pietro A. Della Cristina1, M. Serena Fabbrini2, Aldo Ceriotti2, David Flavell3, Sopsamorn U. Flavell3, Marco Colombatti1
1Dept. Pathology, Sect. Immunology, Policlinico G.B. Rossi, 37134 Verona, Italy; 2Institute of Biology and Agricultural Biotechnology,, CNR, 20133 Milano, Italy; 3Southampton General Hospital, Leukaemia Busters, SO16 6YD Southampton, United Kingdom
Expression of proteins in yeast leads to the secretion of a homogeneous pool of high-quality, fully active molecules, which can be recovered and purified from the culture medium. Pokeweed Antiviral Protein (PAP) was expressed in the P. pastoris as a secreted soluble protein in high quantity and was non toxic to the yeast cells (Rajamohan et al.,1999). We demonstrate that it is possible to use this expression system also for mature saporin (SAP), although with lower yields, for a single-chain variable fragment antibody (scFv) and a recombinant saporin-based IT. Transformants of P. Pastoris were analysed for their ability to secrete recombinant proteins and for growth after induction with methanol. 1) SAP transformants were able to produce and secrete up to 0.3 mg/L of a mature form of SAP, although they grew more slowly than controls. To assess whether SAP was indeed toxic for yeast cells we compared the growth of wt SAP with the growth of the inactive mutant SAP (SAPKQ). We found that SAPKQ mutants grew as fast as the mock-treated controls and produced ten-fold more recombinant protein than wt SAP. 2) The yield of PAP confirms the data of the literature. PAP is non toxic, or less toxic than SAP, to the yeast cells. 3) The amount of the recombinant scFv produced is 10–15 mg/L. 4) The yield of the recombinant IT in Pichia was only 0.4–0.5 mg/L of secreted protein. This can indicate that SAP is toxic to the system also in IT format. We plan to create a system of SAP expression in P. pastoris with an intracellular immunisation strategy to protect cells from the toxic effects of SAP.
M-02
POSTER
Efficient Killing of CD22+ Tumor Cells by a Humanized Diabody-RNase Fusion Protein
Jürgen Krauss1, Michaela Arndt1, Bang Vu2, Andrew Martin3, Huaitian Liu2, Dianne Newton2, Siegfried Seeber1, Susanna Rybak4
1Innere Klinik und Poliklinik (Tumorforschung), Universitätsklinikum Essen, 45122 Essen, Germany, 2National Cancer Institute at Frederick, SAIC, 21701 Frederick, USA, 3Department of Biochemistry and Molecular Biology, University College London, WC1E 6BT London, UK, 4Bionanomics, 32043 Green Cove Springs, USA
CD22-specific immunotoxins are highly potent drugs for the treatment of patients with refractory B-cell non-Hodgkin's lymphoma. However, systemic toxicity and immunogenicity may hamper the wide clinical application of these powerful reagents in cancer patients. To overcome these limitations we have developed a dimeric CD22-specific immunoenzyme capable of simultaneously delivering two RNase effector domains on one molecule to CD22+ tumor cells. For the targeting moiety we first generated a highly stable humanized scFv by grafting the specificity of the clinically established anti-CD22 mAb RFB4 into robust, pre-selected frameworks from a human antibody phage display library. A derived diabody with further engineered interface (VL36Leu→Tyr) retained the same high affinity as the murine anti-CD22 mAb RFB4 (KD = 0.2 nM) as well as full antigen binding after 8 days of incubation in human serum at 37°C. A dimeric immunoenzyme comprising this diabody and Rana pipiens liver ribonuclease I (rapLRI) was generated, expressed as soluble protein in bacteria, and purified to homogeneity. The dimeric fusion protein killed several CD22+ tumor cell lines with high efficacy (IC50 = 3–20 nM) and exhibited 9–48-fold stronger cytotoxicity than a monovalent rapLRI-scFv counterpart. Our results suggest dimeric antibody-ribonuclease fusion proteins as potent immunotherapeutic reagents with presumably large therapeutic indices.
M-03
POSTER
Role of endosomes and Golgi in the endocytosis of the Immunotoxin CD7-saporin
Valeria Giordani1, David Flavell2, Bee Flavell2, Maria Serena Fabbrini3, Rodolfo Ippoliti1
1Basic and Applied Biology, University of L'Aquila, I-67010 L'Aquila, Italy; 2Southampton General Hospital/Leukaemia Busters Charity, SO16 6YD Southampton, United Kingdom; 3Istituto di Biologia e Biotecnologia Agraria, CNR, I-20133 Milano, Italy
Ribosome Inactivating Proteins are enzymes from plants, whose N-glycosidase activity releases single adenine from the 28S rRNA of eukaryotic cells thus inducing irreversible stop of protein synthesis. The most toxic of these enzymes (Ricin) has been taken as a model for the construction of ITs directed toward cancer cells. We have studied the cellular pathway of immunotoxins followed after their internalization in human lymphoma HSB-2 cells over-expressing CD7 antigen. The ITs used are constituted of w.t. saporin (ITSap) and recombinant saporin with KDEL sequence (ITSapKDEL), a retreival sequence for ER transport from the Golgi, chemically linked to monoclonal antibody against CD7 antigen. ITSap (with a fluorescent label) was followed during endocytosis and compared to ricin, by fluorescence microscopy. Cytotoxicity was followed by both colorimetric (WST-1) and radioactive probes (leucine-3H). Brefeldin A (BFA) and NBD C6-ceramide were used to test the localization of toxins at the level of Golgi complex. Chloroquine was used to study the role of endosomal acidification in endocytosis of ITs. Kinetics of internalisation inside the perinuclear zone and partial overlapping suggest that ITSap is transported to the Golgi, but also to other compartments where it could escape to reach the cytoplasm. KDEL sequence seems to influence the cytotoxicity of ITSapKDEL. We suggest that the toxin moiety may be released from compartments different from the ER in the absence of a specific signal peptide, but that the Golgi/ER route may be preferred if the KDEL sequence is present.
M-04
![]()
Identification and characterization of ED-B fibronectin function blocking antibodies
Andreas Menrad1, A. Redlitz1, I. Bahr1, J. Prassler2, Jörg Willuda1, Heike Petrul1
1Corporate Research Oncology, SCHERING AG, 13342 Berlin, Germany; 2MorphoSys AG, 82152 Martinsried, Germany
Introduction:
Fibronectin is an important component of the extracellular matrix that contributes to the maintainance of normal cell morphology and migration. Transformed cells however produce novel isoforms by alternative splicing processes. Here, the ED-B domain seems to be of special importance. ED-B colocalizes only with proliferative endothelium in various tumor samples, whereas normal tissue is not stained as was demonstrated by immunohistochemistry using antibodies directed against ED-B. Despite these compelling results, hardly anything is known about the physiological function of this tightly regulated oncofetal extracellular matrix protein. The high degree of homology between mouse, chicken and human ED-B fibronectin together with the selective expression pattern however point to an important function of this fibronectin splice variant.
Results:
Our investigations demonstrate that recombinant ED-B triggers a receptor-mediated signalling cascade which is essential for attachment, proliferation and matrix invasion/tube formation of human dermal microvascular endothelial cells in vitro. By dissecting the ED-B domain in overlapping peptides we were also able to map the bioactive region to loop 5 of the protein. Furthermore, we could isolate the ED-B receptor by affinity chromatography on immobilized recombinant ED-B. Subsequent MS-analysis identified this ED-B binding protein as the α2β1 integrin, which is involved in the process of angiogenesis.
Based on our new findings we hypothesized that the blockade of the ED-B fibronectin receptor interaction will be detrimental for proliferating endothelial cells. To proof our concept, we successfully identified several high-affinity ED-B-function blocking antibodies (30–80 pM in the monovalent Fab-format) in co-operation with MorphoSys AG. These Fab-fragments were tested in the ED-B expressing murine F9-teratocarcinoma model, where we could clearly demonstrate first signs of efficacy after 5 days of daily i.v. treatment.
Conclusion:
Our data clearly demonstrate that 1) ED-B fibronectin indeed has a novel biological function within the process of angiogenesis and tumor growth and 2) the specific blockade of this pathway leads to an inhibition of tumor growth.
M-05
POSTER
Analysis of different saponins for enhanced specific drug uptake
Christopher Bachran1, Iring Heisler1, Matthias Melzig2, Mark Sutherland1,3, Hendrik Fuchs1
1Institut für Klinische Chemie und Pathobiochemie, Charité – Universitätsmedizin Berlin, Campus Benjamin Franklin, 12200 Berlin, Germany; 2Institut für Pharmazie – Pharmazeutische Biologie, Freie Universität Berlin, 14195 Berlin, Germany; 3Present address: Institute of Cancer Therapeutics, University of Bradford, BD7 3AY Bradford, United Kingdom
The poor cellular uptake and long retention time in the blood is a problem for drugs in targeted tumor therapies. Although the mechanism of action is unknown saponins are able to enhance the uptake of agents into cells. Saponins are a highly diverse group of glycosides containing either a steroidal or triterpenoid aglycon to which one or more sugar chains are attached. The position C4 in the agylcon contains different groups, depending on the saponin. Recently, we described the drastically increased uptake of chimeric toxins into target cells. The chimeric toxins are composed of EGF and the plant toxin saporin. After internalization mediated by receptor-ligand binding, the toxin is delivered to the cytosol resulting in cell death.
Here we compared saponins with different agylcons and different glycosides for their ability to enhance the specific uptake of chimeric toxins. Saponins and chimeric toxin where administered in combination to target cells expressing EGF receptor. All saponins tested resulted in a 10–3000 fold increase in toxicity of the chimeric toxin, indicating an increased internalization. While some saponins promote an unspecific increase in toxicity other reveal a clear target receptor dependent effect. The composition of the different saponins seems to be important for the internalization mechanism. Those saponins containing an aldehyde at position C4 in the aglycon showed the largest increase in cytotoxicity. It may thus be postulated that the aldehyde plays a crucial role in the mechanism of uptake.
M-06
POSTER
Anti-cancer Drug Encapsulation into a Potential Protein-based Tumor Targeted Drug Delivery System
Erdal Ozlu, Mehmet Akif Kilic
Biology, Akdeniz University, 07058 Antalya, Turkey
Introduction:
The ultimate goal in cancer therapy is to develop a targeted system that will selectively destroy cancer cells of the patient. Ferritin proteins form a nanosphere of diameter ~12 nm, with an internal cavity ~8 nm across. Rapidly dividing cancer cells have a high number of ferritin receptors compared to resting cells. For these reason, ferritin is a potential drug encapsulation and carrier system for the targeted delivery of anti-cancer agents to cancer cells.
Results:
Previous studies by our group have shown that doxorubicin can be encapsulated into ferritin nanosphere. In this study, the drug loading capacity of horse ferritin was tested in higher dosages of doxorubicin concentrations (750 and 1500 µM). The recovery of drug-free ferritin was about 80%. In 750 and 1500 µM doxorubicin concentrations, the amount of recovered protein after drug loading was about 65% and 55%, respectively. The amount of drug encapsulated into ferritin nanosphere was found to be higher than 1 ferritin: 5 doxorubicin molecules.
Conclusions:
The study shows that up to 1500 µM doxorubicin concentration, the amount of recovered ferritin does not change dramatically and even in high drug concentration, ferritin can assemble correctly and encapsulate the drug into its internal cavity. Furthermore, when the drug concentration was increased in the reaction mixture, the doxorubicin encapsulation of the ferritin also increases. Therefore, the ferritin-doxorubicin complex could be developed as an anti-cancer drug carrier and consequently, as a targeted drug delivery system for cancer therapy.
M-07
POSTER
Synthesis, Mechanistic Studies and Biological Evaluation of Duocarmycin Analogue Prodrugs for a Selective Treatment of Cancer
Birgit Krewer1, Felix Major1, Marta Zientkowska2, Frauke Alves2, Ingrid Schuberth1, Lutz F. Tietze1
1Institut fuer Organische und Biomolekulare Chemie, Universitaet Goettingen, 37077 Goettingen, Deutschland, 2Abteilung Haematologie und Onkologie, Universitaet Goettingen, 37075 Goettingen, Deutschland
One of the main problems of anticancer therapy is an insufficient differentiation between normal and malignant cells by the known antiproliferant agents, resulting in severe side effects. The antibody directed enzyme prodrug therapy (ADEPT) is a promising development for the selective treatment of cancer, in which a non-toxic prodrug is enzymatically converted into a cytotoxic drug selectively at the surface of malignant cells by an antibody-enzyme conjugate.[1]
New seco-analogues of the highly cytotoxic duocarmycin SA have been developed and detoxified to little toxic glycosidic prodrugs.[2] In vitro cytotoxicity studies reveal an excellent QIC50 value (QIC50: (IC50 Prodrug)/(IC50 Prodrug + corresponding enzyme)) which confirms the suitability of the new prodrugs for ADEPT. The compounds contain a new DNA-binding subunit which not only allows a strong non-covalent bonding to the minor groove of DNA but also improves their water solubility.[3] DNA binding studies using ESI-FTICR mass spectrometry confirm that the new seco-drugs act as highly sequence-specific and efficient DNA alkylating agents.[4] Moreover, we were able to determine the binding position of the monoclonal antibodies by labelling them with fluorescence dyes and detecting them in living mice using an optical imaging system for small animals.
[1] L. F. Tietze, T. Feuerstein, Curr. Pharm. Des. 2003, 9, 2155–2175.
[2] L. F. Tietze, F. Major, I. Schuberth, submitted.
[3] L. F. Tietze, F. Major, Eur. J. Org. Chem. 2006, in press.
[4] L. F. Tietze, B. Krewer, H. Frauendorf, F. Major, I. Schuberth, submitted.
Category: Role of toxin selection
T-01
POSTER
Chimeric Granzyme B Fusion Proteins for Targeted Induction of Apoptosis in Tumor Cells
Benjamin Dälken, Hayat Mahmud, Ulrike Giesübel, Winfried Wels
AG Wels, Georg-Speyer-Haus, 60596 Frankfurt, Germany
The serine protease granzyme B (GrB) of cytotoxic lymphocytes induces apoptosis by direct activation of caspases and cleavage of central caspase substrates. We employed human GrB as an effector function in chimeric fusion proteins that contain the EGFR ligand TGFα or the ErbB2-specific single-chain antibody fragment scFv(FRP5) for selective targeting to tumor cells. GrB-TGFα (GrB-T) and GrB-scFv(FRP5) (GrB-5) proteins expressed in the yeast Pichia pastoris were bifunctional, cleaving GrB substrates, and binding specifically to cells expressing EGFR or ErbB2 target receptors. Upon cell binding the chimeric molecules were internalized into intracellular vesicles, where they showed no cytotoxic activity. However, release of the fusion proteins from these vesicles by the endosomolytic reagent chloroquine resulted in selective tumor cell killing at pico- to nanomolar concentrations of GrB-5 and GrB-T, accompanied by clear signs of apoptotic cell death as early as 3 hours after addition of the proteins. Non-fused GrB expressed in yeast, was also taken up by tumor cells and accumulated in vesicular structures. However, in contrast to the chimeric GrB fusion proteins non-fused GrB could not be liberated to the cytosol by chloroquine, indicating that these vesicular structures are distinct from late endosomes and lysosomes reached after receptor-mediated endocytosis. In contrast, direct cytosolic delivery of GrB with a cationic lipid-based transduction reagent resulted in the induction of apoptotic cell death.
Category: Other
O-01
POSTER
A CD40-CD95L Fusion Protein interferes with CD40L-induced Pro-Survival Signalling and allows Membrane CD40L-Restricted Activation of CD95
Constance Assohou-Luty1, Jeanette Gerspach1, Daniela Siegmund2, Nicole Mueller2, Bertrand Huard3, Gisa Tiegs4, Klaus Pfitzenmaier*1, Harald Wajant*2
1Institute of Cell Biology and Immunology, University of Stuttgart, 70569 Stuttgart, Germany; 2Molekulare Innere Medizin, Medizinische Klinik und Poliklinik II, 97070 Wuerzburg, Germany; 3Dermatology Department, Geneva University Medical Center, 1211 Geneva 4, Switzerland; 4Institute of Experimental and Clinical Pharmacology and Toxicology, University of Erlangen-Nuremberg, 91054 Erlangen, Germany
We analyzed a novel bifunctional fusion protein, CD40ed-CD95Led, consisting amino-terminally of the extracellular domain of CD40 and carboxy-terminally of the extracellular domain of CD95L. On cells expressing no CD40L this fusion protein is poorly active with respect to CD95 activation (ED50 > 1 µg/ml), but it stimulates CD95 signaling with high efficiency after binding to CD40L (ED50 < 1 ng/ml). Thus, cell surface immobilization mediated by the CD40 part of the molecule unmasks the high latent CD95-stimulating capacity of the otherwise poorly active CD95L fusion protein. Moreover, interaction of the CD40 part of CD40ed-CD95Led with CD40L prevents activation of cellular CD40. The CD40ed-CD95Led fusion protein therefore simultaneously blocks anti-apoptotic CD40 activation and induces CD95-mediated apoptosis. Indeed, T47D cells displaying an antiapoptotic autocrine CD40-CD40L signalling loop were significantly more sensitive towards CD40ed-CD95Led than towards soluble CD95L artificially activated by crosslinking. Fusion proteins of RANK and CD95L (RANKed-CD95Led) and CD40 and TRAIL (CD40ed-TRAILed) having a similar domain architecture as CD40ed-CD95Led displayed RANKL-dependent CD95 activation and CD40L-dependent TRAILR2 activation. Thus, fusion proteins of the presented structure may also be constructed to combine other beneficial pairs of the TNF ligand and receptor family into one molecule.
ed: extracellular domain; TRAIL: tumor necrosis factor-related apoptosis inducing ligand
O-02
POSTER
Adenovirus-based vaccine for prevention and therapy of HPV-associated cervical cancer
Corinna Hoffmann
Charité, 12203 Berlin, Germany
Cervical cancer belongs to the most common cancers among women worldwide. About 80% of cervical cancer cases are associated with infection by HPV type 16 and 18. Malignant transformation of cervical cells occurs after a latent phase of persistent infection due to chromosomal integration and overexpression of HPV oncogenes E6 and E7. Constitutive expression of viral oncogenes in malignant cells makes them a favourable target for immunotherapeutic strategies. Currently, prophylactic vaccines have been developed, targeting late capsid proteins L1 and L2. So far there are no therapeutic vaccines available that induce effective cellular immunity against HPV-positive cancer cells and its precursor lesions.
We designed a recombinant fusion gene that consists of 14 fragments of HPV16/18 E6/7. To reduce the transforming activity of E6 and E7, fragments with oncogenic activity were deleted whereas antigenic epitopes were preserved. These antigenic fragments were rearranged and fused to build the p14 recombinant gene. To increase immunological efficiency the recombinant gene was cloned into a replication-deficient adenovirus. The viral backbone ensures high transduction rates, high in vivo expression of the fusion gene, and increases immune responses arising from its intrinsic antigenicity (adjuvant effect). In animal tumour models vaccination resulted in complete and permanent remission of transplanted HPV16 E6/7 expressing tumour cells. Further experiments should reveal the potential of this vaccine in human immune cells of cervical cancer patients.
POSTER and SHORT COMMUNICATION: Thursday 30 March 2006, 18:22
Ep-CAM-directed liposomal drug delivery systems for targeted cancer therapy
Sajid Hussain1, 2, Andreas Pluckthun1, Theresa Allen3, Uwe Zangemeister-Wittke4
1Biochemistry Institute, University of Zurich, 8057 Zurich, Switzerland; 2Department of Oncology, University Hospital of Zurich, 8044 Zurich, Switzerland; 3Department of Pharmacology, University of Alberta, T6G 2H7 Edmonton, Canada; 4Department of Pharmacology, University of Bern, 3010 Bern, Switzerland
Site-directed delivery of anti-cancer agents to tumors increases their therapeutic efficacy and reduces toxicity to normal tissues. Nanovectors, such as sterically stabilized immunoliposomes (SILs) that specifically bind to internalizing tumor antigens, offer the promise of enhanced safety and delivery of large quantities of anti-cancer agents to tumors. The epithelial cell adhesion molecule (Ep-CAM) is abundantly expressed in solid tumors, but shows limited distribution in normal epithelial tissues. To generate Ep-CAM-directed immunoliposomes for targeted cancer therapy, the humanized high affinity scFv 4D5MOCB was covalently coupled to the surface of sterically stabilized liposomes containing either the Bcl-2/Bcl-xL bispecific antisense oligonucleotide 4625 (SIL-25) or the anthracyline doxorubicin (SIL-Dox). Both immunoliposome preparations showed rapid and specific binding to Ep-CAM-positive tumor cells, which was 10 to 20-fold higher than non-targeted liposomes (SL-25 or SL-Dox). SIL-25 effectively downregulated Bcl-2 and Bcl-xL expression, facilitated apoptosis and enhanced chemosensitivity. SIL-Dox were significantly more cytotoxic than the non-targeted SL-Dox. In vivo pharmacokinetic and biodistribution analysis of SIL-Dox showed long circulation times and effective tumor accumulation. They also demonstrated enhanced anti-tumor activity against established tumor xenografts compared to the free drug or non-targeted SL-Dox. Our data suggest the use of Ep-CAM-directed nanovectors, such as antisense or drug-loaded immunoliposomes, for targeted therapy of solid tumors.
O-04
COMPANY POSTER
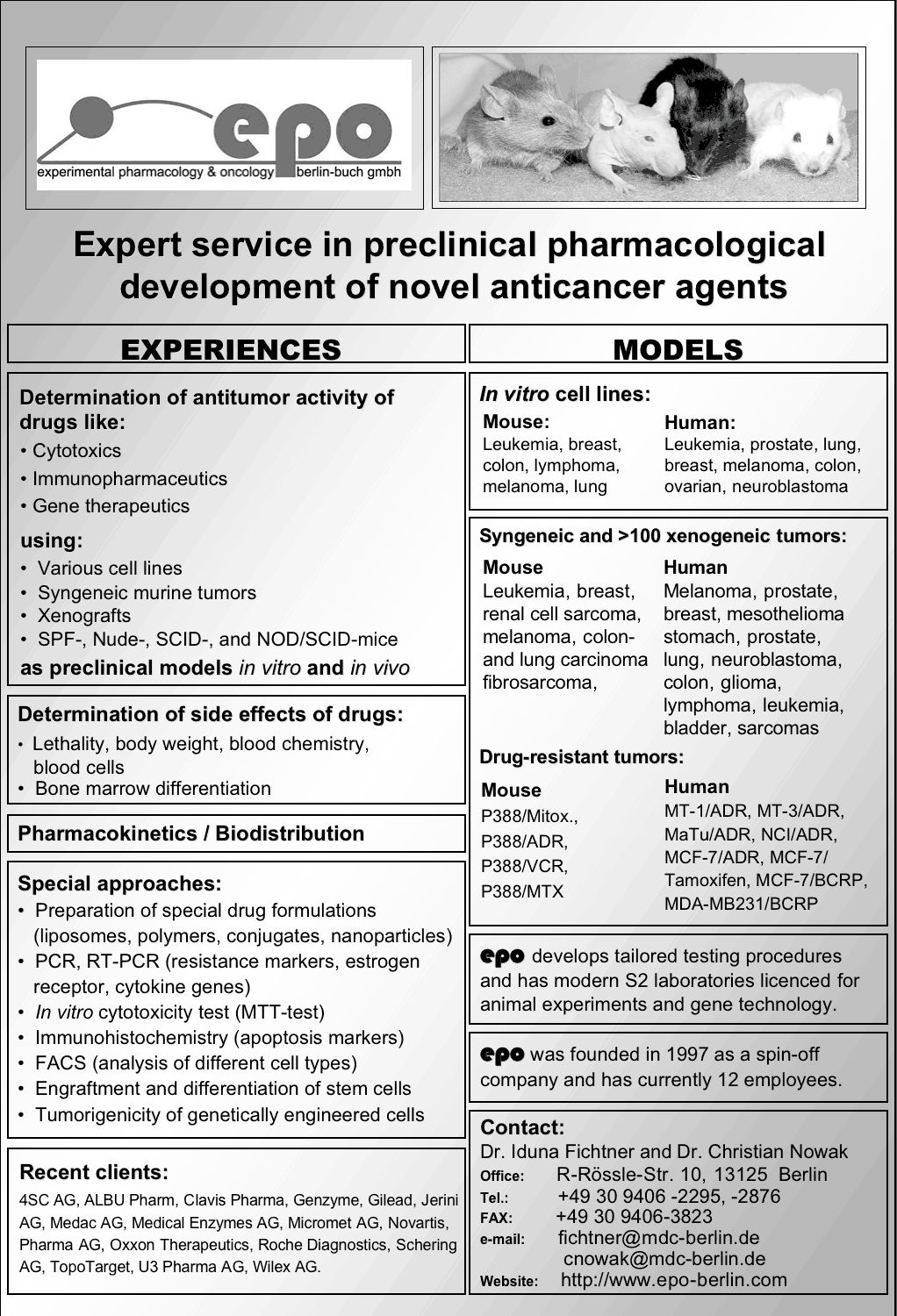
EPO Berlin-Buch GmbH - Company profile
Reiner Zeisig
EPO Berlin-Buch GmbH, 13125 Berlin, Germany
O-05
POSTER
Synthesis and evaluation of Ga-68 labeled Y2-selective peptides for in vivo receptor targeting using small animal PET
Denise Zwanziger1, Irfan U. Kahn1, Jörn Schlesinger2, Ralf Bergmann2, Annette G. Beck-Sickinger1
1Institute of Biochemistry, University of Leipzig, D-04103 Leipzig, Germany; 2Institute of Radiopharmacy, Research Centre Rossendorf, 01328 Dresden, Germany
Peptide YY (PYY) is a 36 amino acid peptide amide that belongs to the PP-hormone family. It selectively binds to at least two G-protein coupled receptors named Y2- and Y5-receptor. These receptors are overexpressed in various tumors like neuroblastomas and nephroblastomas. With regard to clinical applications, NPY2 and NPY5 receptors may act as in vivo targets for receptor-directed therapy and diagnosis of the tumors, however there are specific and in vivo stabile ligands still required. The aim of the work was to synthesize receptor ligands, which could be radioloabeled with the positron emitting nuclides Ga-68 for small animal positron emission tomography (PET).
PYY derivatives were synthesized by solid-phase peptide synthesis, characterized by HPLC/MALDI techniques, purified by preparative HPLC and linked to DOTA (1,4,7,10-tetraazacyclotetradecane-N, N´,N´´,N´´´tetraacetic acid) as chelator for the 68Ga(III). The in vitro binding affinity of the peptides was studied in SMS-KAN cells expressing NPY2 receptors by competitive receptor binding assays. Two Ga-68 labeled derivatives were studied in vivo by small animal PET in rats and mice. The activity was fast eliminated into the urine. One hour after injection was only in the kidneys remaining activity detected.
Optimized Y2-selective and in vivo stabile peptides will be developed and tested for tumor targeting.
SHORT COMMUNICATION: Friday 31 March 2006, 12:32
Dendritic Polymers for DNA and Drug-Delivery
Rainer Haag
Fachbereich Biologie, Chemie, Pharmazie, Freie Universität Berlin, Institut für Chemie und Biochemie, 14195 Berlin, Germany
E-mail: haag@chemie.fu-berlin.de, Internet: www.polytree.de
The supramolecular core-shell architectures presented here, are based on a dendritic (“tree-like”) core molecule, surrounded by a covalently attached shell, which determines the physical and biological properties of the nanocarrier.[1]
For in vivo applications, those nanocarriers will have to be water-soluble and non-toxic. The key step of the drug-delivery process is the uptake of loaded nanocarriers into the cell. In the case of oligonucleotide transfection, uptake is provided by enhanced endocytosis using polycationic dendritic polymers.[2] For the uptake of anti-tumor drugs, entry of the nanocarriers in the tumor tissue can be further enhanced due to the permeability of tumor blood vessels.[1] Therefore, we are interested in the design of “universal” nanocarriers, for selective drug and oligonucleotide release based on hyperbranched polymers, such as polyglycerol and polyethyleneimine as core-molecules.[3] In addition, pH-controlled or enzymatic release upon an external stimulus is another feature of these dendritic nanocarriers we are investigating.[3]
[1] Haag, R.; Kratz, F. Angew. Chem. Int. Ed. 2006, 45, 1198
[2] Krämer M., Haag R. et al., Chem. Bio. Chem. 2004, 5, 1081
[3] Krämer M., Haag R. et al. Angew. Chem. Int. Ed. 2002, 41, 4252
O-07
POSTER
Development of Prodrugs Selectively Activated by Tumour Expressed Cytochrome P450s
Mark Sutherland1, Klaus Pors1, Nicola Harris1, Mark Searcey2, Meritxell Guino2, Laurence Patterson1
1Institute of Cancer Therapeutics, University of Bradford, BD7 3AY Bradford, United Kingdom, 2School of Pharmacy, University of London, WC1N 1AX London, United Kingdom
Cytochrome P450s (CYP's) are a superfamily of mixed function oxidases responsible for metabolising xenobiotics, steroids and eicosanoids. Metabolism by the CYP 1-3 family is generally viewed as a route to drug detoxification and increased elimination. There is considerable evidence demonstrating expression of a wide range of CYPs in all the major clinically derived solid tumours and even over-expression of selected isoforms. Pyrolloindoles are a family of natural agents that interfere with DNA replication and induce cell death, however, they are not tumour selective. We have synthesised prodrug analogues lacking the hydroxyl group that initiates DNA binding. Hydroxylation by CYPs, one of the major reactions catalysed, activates the prodrug. The prodrug DP-7, has been tested in vitro on CHO cells and transfectants over-expressing CYP1A1 and CYP1B1, two extrahepatic CYPs. The IC50s were greater than 50, 0.1 and 5 µM respectively. This activity is similar to that obtained from DP-7 activation by bactosomes containing either CYP1A1 or CYP1B1. This suggests that the prodrug is predominantly metabolised by CYP1A1. This is further supported by HPLC data that reveals that CYP1A1 bactosomes metabolise DP-7 to a greater extent than CYP1B1 bactosomes. To exclude possible hepatic damage, DP-7 was incubated in the presence of mouse liver homogenates. The low level of metabolites suggests that DP-7 is not readily hydroxylated by all CYPs and may preclude liver activation and elimination, which is a significant finding since the CYPs are expressed at high levels in the liver.
![]()
Identification of tumor endothelial antigens by means of tissue panning using an antibody phage display library
Yong-Jiang Cao, Andreas Menrad, Jörg Willuda, Heike Petrul, V. Badock, K. Bosslet
Antiangiogenesis Research, SCHERING AG, 13342 Berlin, Germany
Objectives: Tumor angiogenesis is a complex process in which the phenotype of the proliferating endothelial cells is strongly controlled by the tumor's microenvironment. Once endothelial cells are isolated and grown under cell culture conditions they undergo dramatic phenotypic changes (“phenotypic drift”). Such in vitro artifacts have to be avoided or minimized so that cell surface structures relevant for the process of tumor angiogenesis can be identified. In this work we described a novel methodology to scan cell surface structures from freshly isolated human tumor tissues for target identification using an antibody phage display library.
Methods: We have performed antibody phage display by cell/tissue panning strategies. Single cell suspensions from human tumor tissues were prepared by mild enzymatic digestion. An antibody phage display library (diversity: 5.2 x 109) were added and panned. Phages bound tumor endothelial cells were separated from irrelevant contaminating cells by means of magnetic bead selection. To identify specific scFv antibodies for tumor endothelial cells, eluted phage-antibodies were screened by ELISA on untreated tumor-derived endothelial cells and irrelevant cells as a negative control. To demonstrate specificity of phages for tumor vasculature, all ELISA positive antibodies were further characterized by phage-mab-immunohistochemistry on human tumor tissues and benign tissue.
Results: We have successfully established a novel selection methodology to scan cell surface structures from human tumor tissues. Several (tumor-) endothelium-specific antibodies were isolated from the scFv phage display library. The specificity was demonstrated by immunohistochemisty. With a selected soluble scFv antibody we could isolate the respective antigen from a tumor endothelial cell lysate. It was identified as endoglin (CD105) which represents a well-described molecule associated with the process of tumor angiogenesis.
Conclusion: Cell- and tissue panning strategies provide a powerful tool for the identification of targets which are expressed during (tumor)angiogenesis.
Design, Synthesis and Evaluation of Novel Anticancer Agents or Nanodevices towards Inhibition and/or Imaging of Tumour Neoangiogenesis
Gérard Déléris1, Andreas Bikfalvi2, Karine Estieu-Gionnet1, 2, Mario Goncalves1, Thomas Berthelot1, Marie-Claude Clochard3, Olivia Cuscito3
1Bio-Organic Chemistry, CNRS, University Bordeaux 2, 33076 Bordeaux, France, 2Université Bordeaux 1, INSERM E0113, Molecular Mechanisns of Angiogenesis, 33 405 Talence, Farnce, 3DSM/LPI, CEA Saclay, 91 191 Gif/ Yvette, France
Angiogenesis modulating and targeting is of outstanding importance for its inhibition (and imaging) in an anticancer approach. Angiogenic key factor is, VEGF, a 165 amino-acids peptide which promotes angiogenesis through dimerization of VEGF-R2. Structural studies highlighted that a basic, looplike,sequence is responsible for the activity. We designed and synthesized a series of cyclic peptides mimicking this loop. When tested for their ability to displace binding of labelled VEGF to its receptor several cyclopeptides exhibited a micromolar inhibitory activity. They also inhibit proliferation and migration of endothelial cells and were potent inhibitors of the growth or human glioma established intracranially at mouse, leading to an important increase in survival of the treated animals (1-3). Specific delivery of antiangiogenesis nano-devices is also an important challenge for therapy and imaging. For this purpose, cyclopeptide carriers have been chemically coupled to imaging agents or polymers (4).
References:
1. Bello, L., Deleris, G. and Bikfalvi, A. et al. Clin Cancer Res 10 (13) 4527-37 (2004)
2. Zilberberg, L., Shinkaru, Deleris, G., Bikfalvi, A. et al. J Biol Chem 278 (37) 35564-73 (2003)
3. Shinkaruk, S., Bayle, M., Lain, G. and Deleris, G. Curr Med Chem Anti-Canc Agents 3 (2) 95-117 (2003)
4. Goncalves, M., Estieu-Gionnet, K., Berthelot, T., Lain, G., Bayle, M., Canron, X., Betz, N., Bikfalvi, A. and Deleris, G. Pharm Res 22 (8) 1411-21 (2005)